

Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl-CoA synthetase 1
- PMID: 22308341
- PMCID: PMC3311324
- DOI: 10.1073/pnas.1111600109
Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl-CoA synthetase 1
Abstract
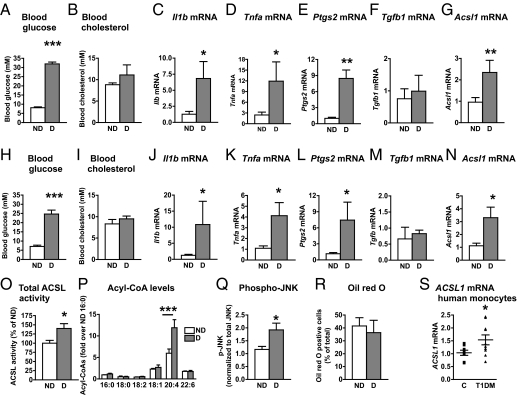
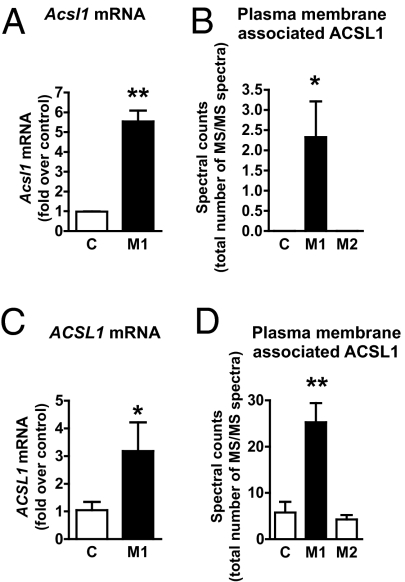
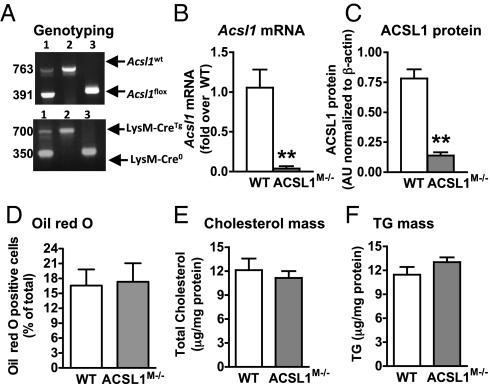
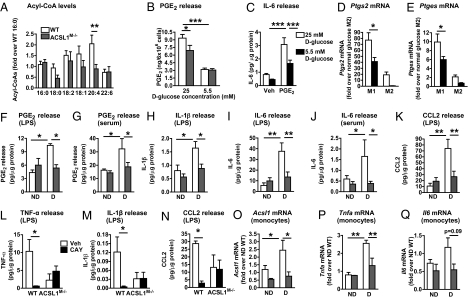
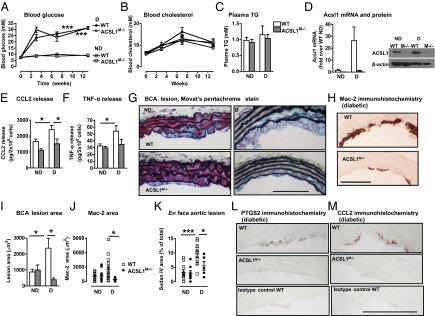
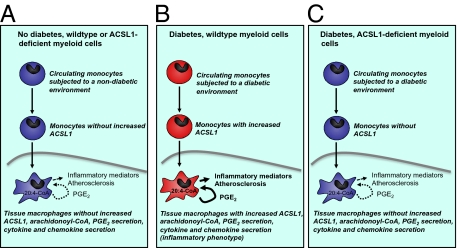
The mechanisms that promote an inflammatory environment and accelerated atherosclerosis in diabetes are poorly understood. We show that macrophages isolated from two different mouse models of type 1 diabetes exhibit an inflammatory phenotype. This inflammatory phenotype associates with increased expression of long-chain acyl-CoA synthetase 1 (ACSL1), an enzyme that catalyzes the thioesterification of fatty acids. Monocytes from humans and mice with type 1 diabetes also exhibit increased ACSL1. Furthermore, myeloid-selective deletion of ACSL1 protects monocytes and macrophages from the inflammatory effects of diabetes. Strikingly, myeloid-selective deletion of ACSL1 also prevents accelerated atherosclerosis in diabetic mice without affecting lesions in nondiabetic mice. Our observations indicate that ACSL1 plays a critical role by promoting the inflammatory phenotype of macrophages associated with type 1 diabetes; they also raise the possibilities that diabetic atherosclerosis has an etiology that is, at least in part, distinct from the etiology of nondiabetic vascular disease and that this difference is because of increased monocyte and macrophage ACSL1 expression.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Comment in
-
Promoting atherosclerosis in type 1 diabetes through the selective activation of arachidonic acid and PGE(2) production.Circ Res. 2012 Aug 3;111(4):394-6. doi: 10.1161/CIRCRESAHA.112.273508. Circ Res. 2012. PMID: 22859668
References
-
- Devaraj S, et al. Increased monocytic activity and biomarkers of inflammation in patients with type 1 diabetes. Diabetes. 2006;55:774–779. - PubMed
-
- Shanmugam N, Gaw Gonzalo IT, Natarajan R. Molecular mechanisms of high glucose-induced cyclooxygenase-2 expression in monocytes. Diabetes. 2004;53:795–802. - PubMed
-
- Natarajan R, Nadler JL. Lipid inflammatory mediators in diabetic vascular disease. Arterioscler Thromb Vasc Biol. 2004;24:1542–1548. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P01 HL018645/HL/NHLBI NIH HHS/United States
- DK59935/DK/NIDDK NIH HHS/United States
- P30DK056350/DK/NIDDK NIH HHS/United States
- DK082841/DK/NIDDK NIH HHS/United States
- R01 HL062887/HL/NHLBI NIH HHS/United States
- R24 DK082841/DK/NIDDK NIH HHS/United States
- P30DK089507/DK/NIDDK NIH HHS/United States
- HL018645/HL/NHLBI NIH HHS/United States
- P30 DK035816/DK/NIDDK NIH HHS/United States
- P30 DK056350/DK/NIDDK NIH HHS/United States
- P30 DK020572/DK/NIDDK NIH HHS/United States
- HL062887/HL/NHLBI NIH HHS/United States
- P30 DK17047/DK/NIDDK NIH HHS/United States
- DK07448/DK/NIDDK NIH HHS/United States
- HL092969/HL/NHLBI NIH HHS/United States
- R01 HL097365/HL/NHLBI NIH HHS/United States
- P30 DK089507/DK/NIDDK NIH HHS/United States
- T32 HL007828/HL/NHLBI NIH HHS/United States
- HL097365/HL/NHLBI NIH HHS/United States
- P30 DK089503/DK/NIDDK NIH HHS/United States
- R01 DK059935/DK/NIDDK NIH HHS/United States
- P01 HL092969/HL/NHLBI NIH HHS/United States
- T32 HL07828/HL/NHLBI NIH HHS/United States
- P30 DK017047/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
