

Genome-wide scans provide evidence for positive selection of genes implicated in Lassa fever
- PMID: 22312054
- PMCID: PMC3267117
- DOI: 10.1098/rstb.2011.0299
Genome-wide scans provide evidence for positive selection of genes implicated in Lassa fever
Abstract
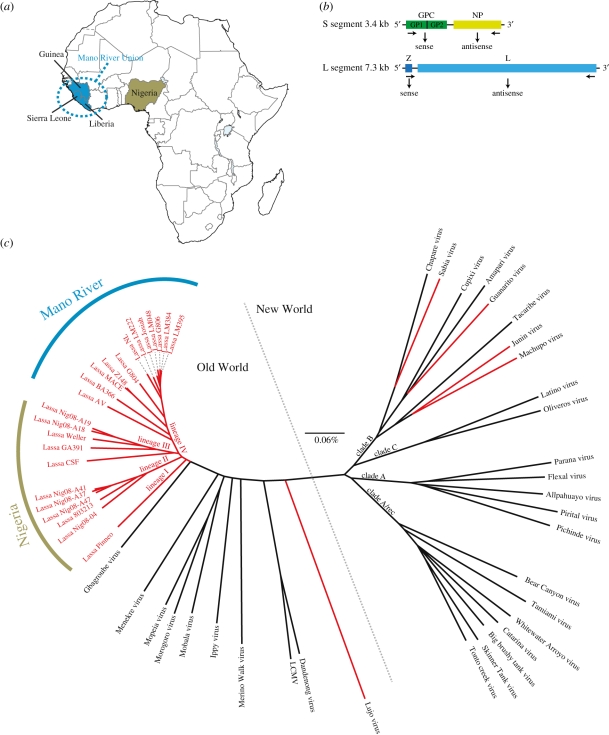
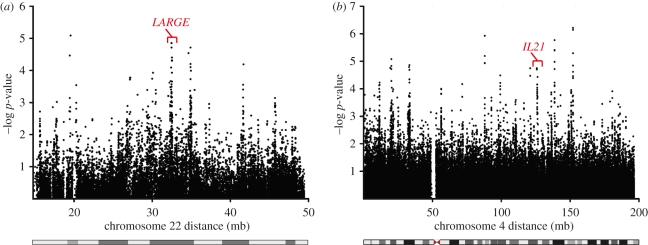
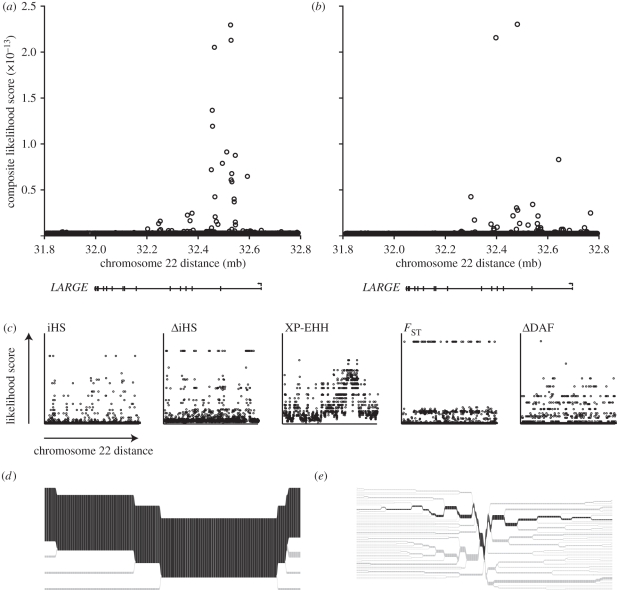
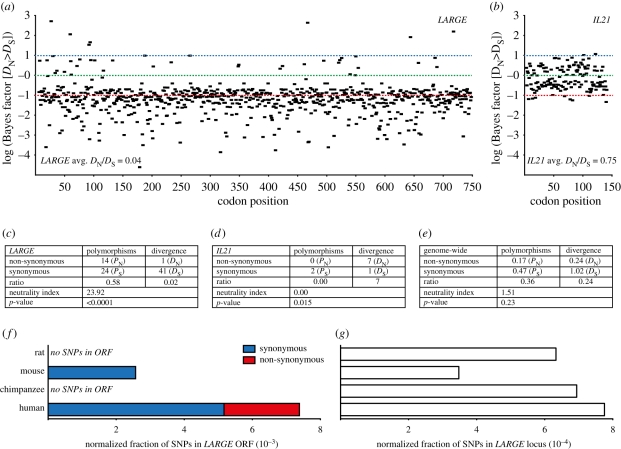
Rapidly evolving viruses and other pathogens can have an immense impact on human evolution as natural selection acts to increase the prevalence of genetic variants providing resistance to disease. With the emergence of large datasets of human genetic variation, we can search for signatures of natural selection in the human genome driven by such disease-causing microorganisms. Based on this approach, we have previously hypothesized that Lassa virus (LASV) may have been a driver of natural selection in West African populations where Lassa haemorrhagic fever is endemic. In this study, we provide further evidence for this notion. By applying tests for selection to genome-wide data from the International Haplotype Map Consortium and the 1000 Genomes Consortium, we demonstrate evidence for positive selection in LARGE and interleukin 21 (IL21), two genes implicated in LASV infectivity and immunity. We further localized the signals of selection, using the recently developed composite of multiple signals method, to introns and putative regulatory regions of those genes. Our results suggest that natural selection may have targeted variants giving rise to alternative splicing or differential gene expression of LARGE and IL21. Overall, our study supports the hypothesis that selective pressures imposed by LASV may have led to the emergence of particular alleles conferring resistance to Lassa fever, and opens up new avenues of research pursuit.
Figures
References
-
- Haldane J. B. S. 1949. Disease and evolution. Ric. Sci. Suppl. A. 19, 68–76
-
- Allison A. C. 1954. Protection afforded by sickle-cell trait against subtertian malarial infection. BMJ 1, 290–29410.1136/bmj.1.4857.290 (doi:10.1136/bmj.1.4857.290) - DOI - DOI - PMC - PubMed
-
- Sabeti P. C., et al. 2006. Positive natural selection in the human lineage. Science 312, 1614–162010.1126/science.1124309 (doi:10.1126/science.1124309) - DOI - DOI - PubMed
-
- Sabeti P. C., et al. 2002. Detecting recent positive selection in the human genome from haplotype structure. Nature 419, 832–83710.1038/nature01140 (doi:10.1038/nature01140) - DOI - DOI - PubMed
-
- The International HapMap Consortium. 2003. The International HapMap Project. Nature 426, 789–79610.1038/nature02168 (doi:10.1038/nature02168) - DOI - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
