

From laptop to benchtop to bedside: structure-based drug design on protein targets
- PMID: 22316152
- PMCID: PMC3820560
- DOI: 10.2174/138161212799436386
From laptop to benchtop to bedside: structure-based drug design on protein targets
Abstract
As an important aspect of computer-aided drug design, structure-based drug design brought a new horizon to pharmaceutical development. This in silico method permeates all aspects of drug discovery today, including lead identification, lead optimization, ADMET prediction and drug repurposing. Structure-based drug design has resulted in fruitful successes drug discovery targeting proteinligand and protein-protein interactions. Meanwhile, challenges, noted by low accuracy and combinatoric issues, may also cause failures. In this review, state-of-the-art techniques for protein modeling (e.g. structure prediction, modeling protein flexibility, etc.), hit identification/ optimization (e.g. molecular docking, focused library design, fragment-based design, molecular dynamic, etc.), and polypharmacology design will be discussed. We will explore how structure-based techniques can facilitate the drug discovery process and interplay with other experimental approaches.
Conflict of interest statement
The authors declare that they do not have competing interests.
Figures
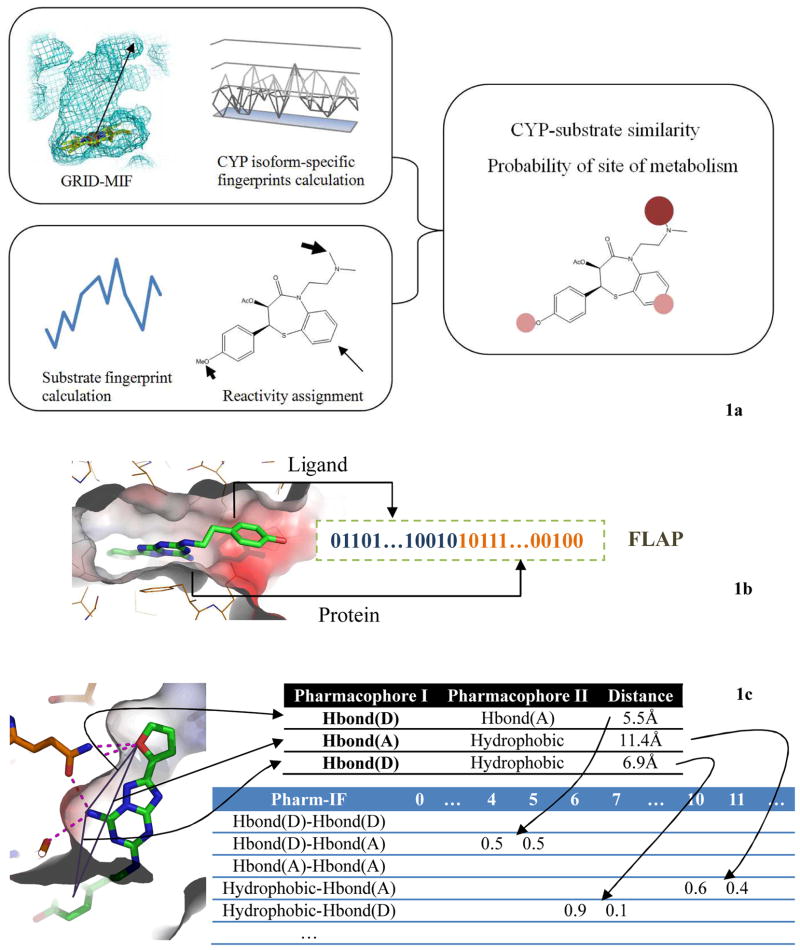
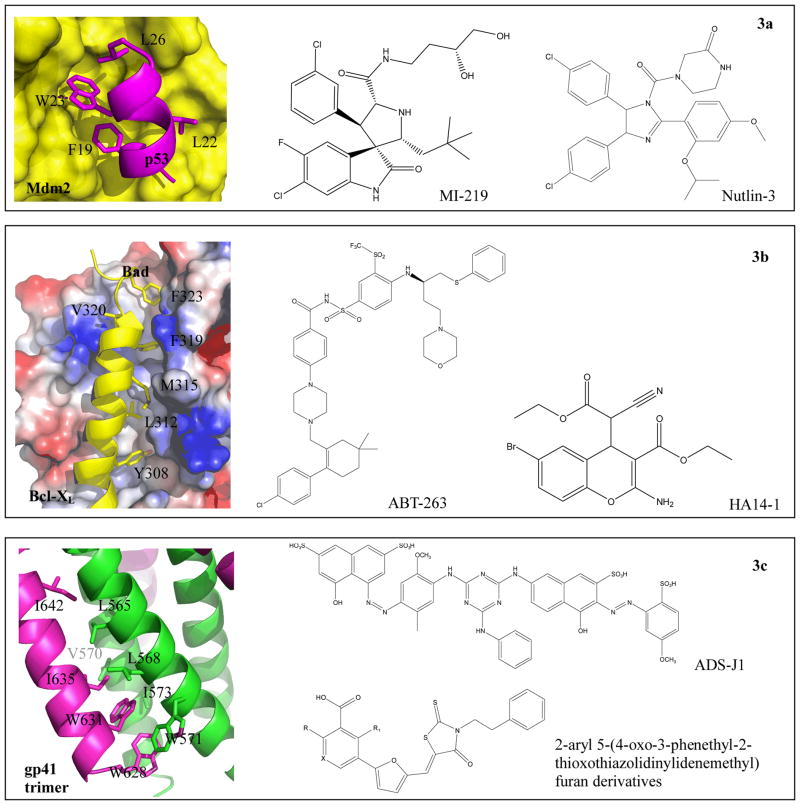
References
-
- Zhang S. Structure/Ligand-based Drug Design and Structure Bioinformatics: Basics, Concepts, Methods, and Applications. VDM Verlag; 2009.
-
- Jorgensen WL. The many roles of computation in drug discovery. Science. 2004 Mar 19;303(5665):1813–8. - PubMed
-
- Weigelt J. Structural genomics-impact on biomedicine and drug discovery. Exp Cell Res. May 1;316(8):1332–8. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
