

msCompare: a framework for quantitative analysis of label-free LC-MS data for comparative candidate biomarker studies
- PMID: 22318370
- PMCID: PMC3433919
- DOI: 10.1074/mcp.M111.015974
msCompare: a framework for quantitative analysis of label-free LC-MS data for comparative candidate biomarker studies
Abstract
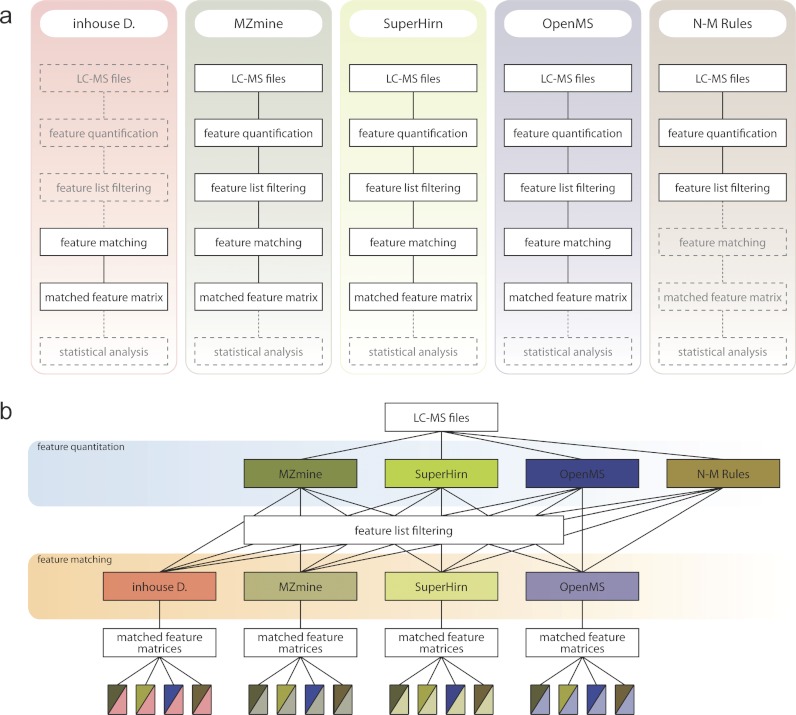
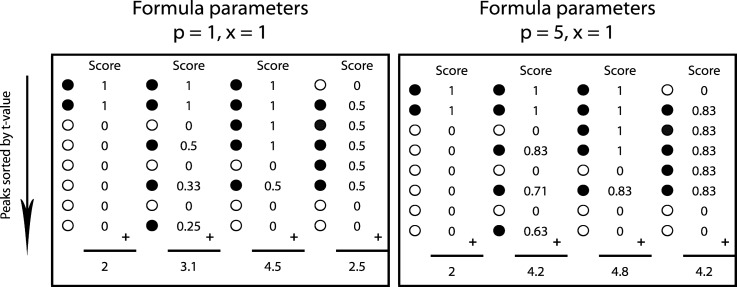
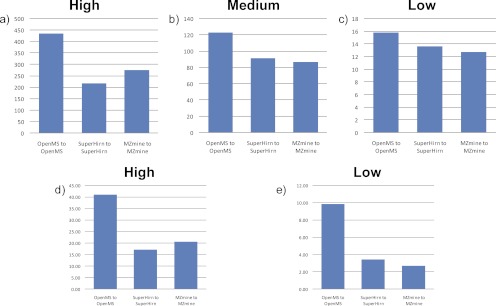
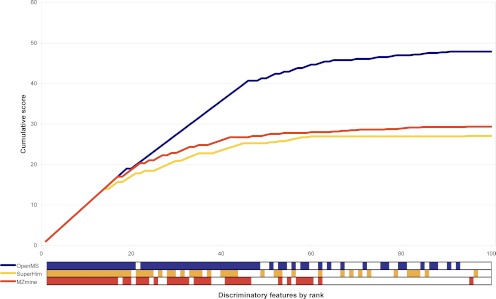
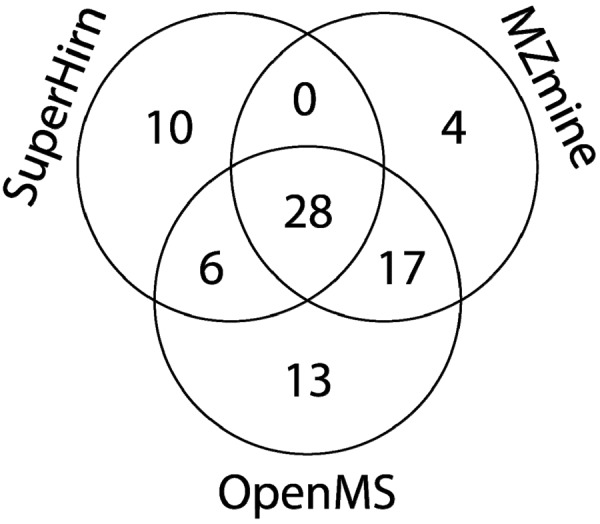
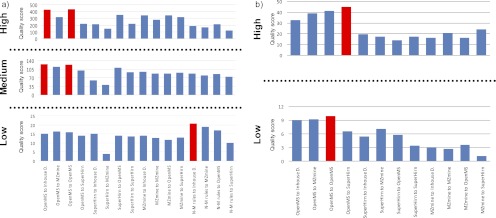
Data processing forms an integral part of biomarker discovery and contributes significantly to the ultimate result. To compare and evaluate various publicly available open source label-free data processing workflows, we developed msCompare, a modular framework that allows the arbitrary combination of different feature detection/quantification and alignment/matching algorithms in conjunction with a novel scoring method to evaluate their overall performance. We used msCompare to assess the performance of workflows built from modules of publicly available data processing packages such as SuperHirn, OpenMS, and MZmine and our in-house developed modules on peptide-spiked urine and trypsin-digested cerebrospinal fluid (CSF) samples. We found that the quality of results varied greatly among workflows, and interestingly, heterogeneous combinations of algorithms often performed better than the homogenous workflows. Our scoring method showed that the union of feature matrices of different workflows outperformed the original homogenous workflows in some cases. msCompare is open source software (https://trac.nbic.nl/mscompare), and we provide a web-based data processing service for our framework by integration into the Galaxy server of the Netherlands Bioinformatics Center (http://galaxy.nbic.nl/galaxy) to allow scientists to determine which combination of modules provides the most accurate processing for their particular LC-MS data sets.
Figures
References
-
- Chen G., Pramanik B. N. (2009) Application of LC/MS to proteomics studies: Current status and future prospects. Drug Discov. Today 14, 465–471 - PubMed
-
- Nilsson T., Mann M., Aebersold R., Yates J. R., 3rd, Bairoch A., Bergeron J. J. (2010) Mass spectrometry in high-throughput proteomics: Ready for the big time. Nat. Methods 7, 681–685 - PubMed
-
- Domon B., Aebersold R. (2010) Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 28, 710–721 - PubMed
-
- Allwood J. W., Goodacre R. (2010) An introduction to liquid chromatography-mass spectrometry instrumentation applied in plant metabolomic analyses. Phytochem. Anal. 21, 33–47 - PubMed
-
- Griffiths W. J., Wang Y. (2009) Mass spectrometry: From proteomics to metabolomics and lipidomics. Chem. Soc. Rev. 38, 1882–1896 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
