

Small molecule antagonist reveals seizure-induced mediation of neuronal injury by prostaglandin E2 receptor subtype EP2
- PMID: 22323596
- PMCID: PMC3286971
- DOI: 10.1073/pnas.1120195109
Small molecule antagonist reveals seizure-induced mediation of neuronal injury by prostaglandin E2 receptor subtype EP2
Abstract
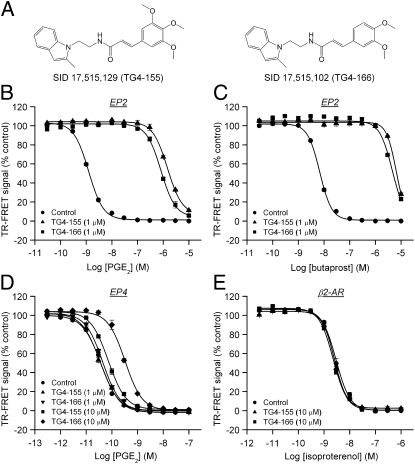
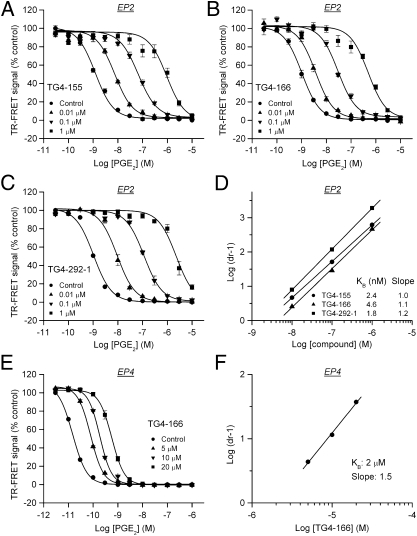
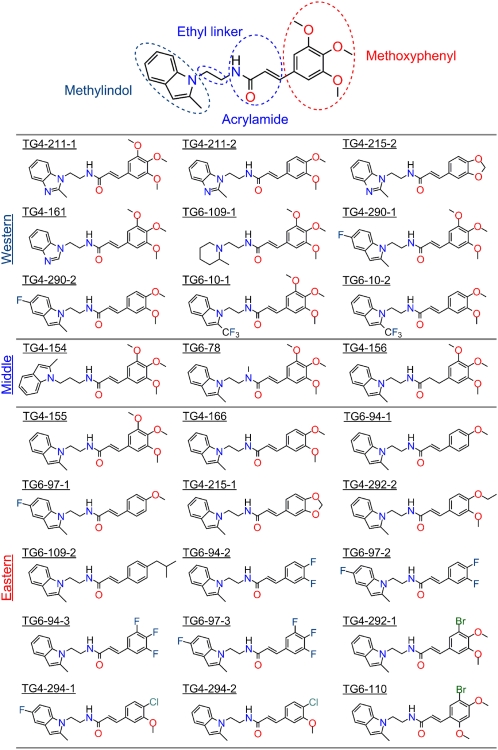
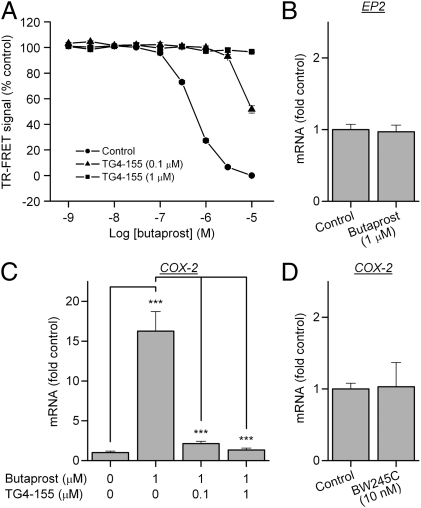
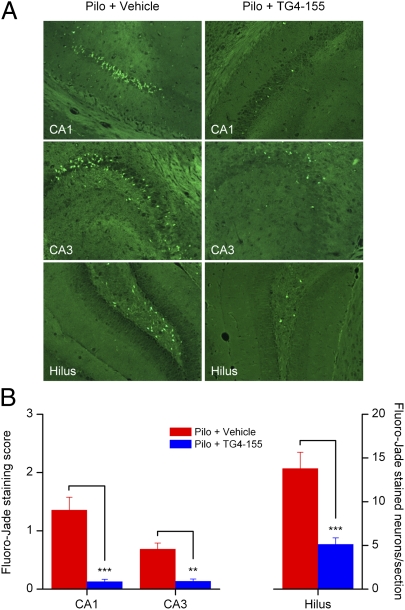
With interest waning in the use of cyclooxygenase-2 (COX-2) inhibitors for inflammatory disease, prostaglandin receptors provide alternative targets for the treatment of COX-2-mediated pathological conditions in both the periphery and the central nervous system. Activation of prostaglandin E2 receptor (PGE(2)) subtype EP2 promotes inflammation and is just beginning to be explored as a therapeutic target. To better understand physiological and pathological functions of the prostaglandin EP2 receptor, we developed a suite of small molecules with a 3-aryl-acrylamide scaffold as selective EP2 antagonists. The 12 most potent compounds displayed competitive antagonism of the human EP2 receptor with K(B) 2-20 nM in Schild regression analysis and 268- to 4,730-fold selectivity over the prostaglandin EP4 receptor. A brain-permeant compound completely suppressed the up-regulation of COX-2 mRNA in rat cultured microglia by EP2 activation and significantly reduced neuronal injury in hippocampus when administered in mice beginning 1 h after termination of pilocarpine-induced status epilepticus. The salutary actions of this novel group of antagonists raise the possibility that selective block of EP2 signaling via small molecules can be an innovative therapeutic strategy for inflammation-related brain injury.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Marcheselli VL, Bazan NG. Sustained induction of prostaglandin endoperoxide synthase-2 by seizures in hippocampus. Inhibition by a platelet-activating factor antagonist. J Biol Chem. 1996;271:24794–24799. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
