

Induction and function of type I and III interferon in response to viral infection
- PMID: 22323926
- PMCID: PMC3272644
- DOI: 10.1016/j.coviro.2011.11.001
Induction and function of type I and III interferon in response to viral infection
Abstract
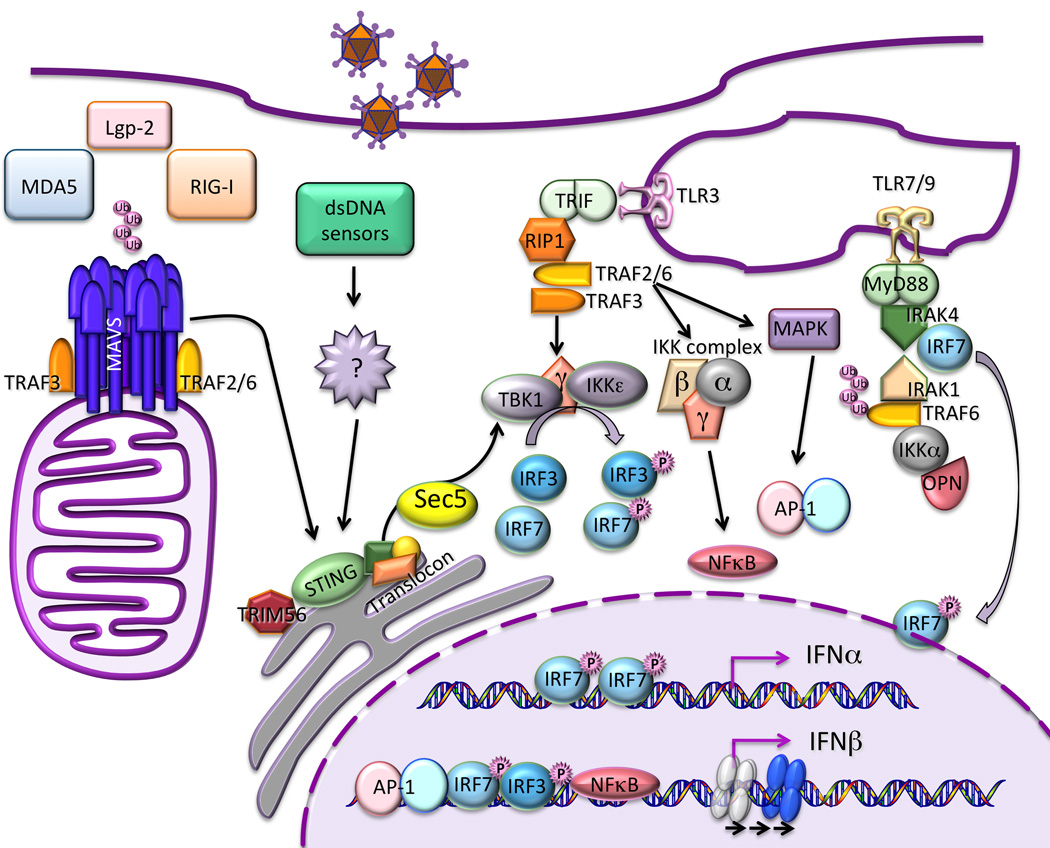
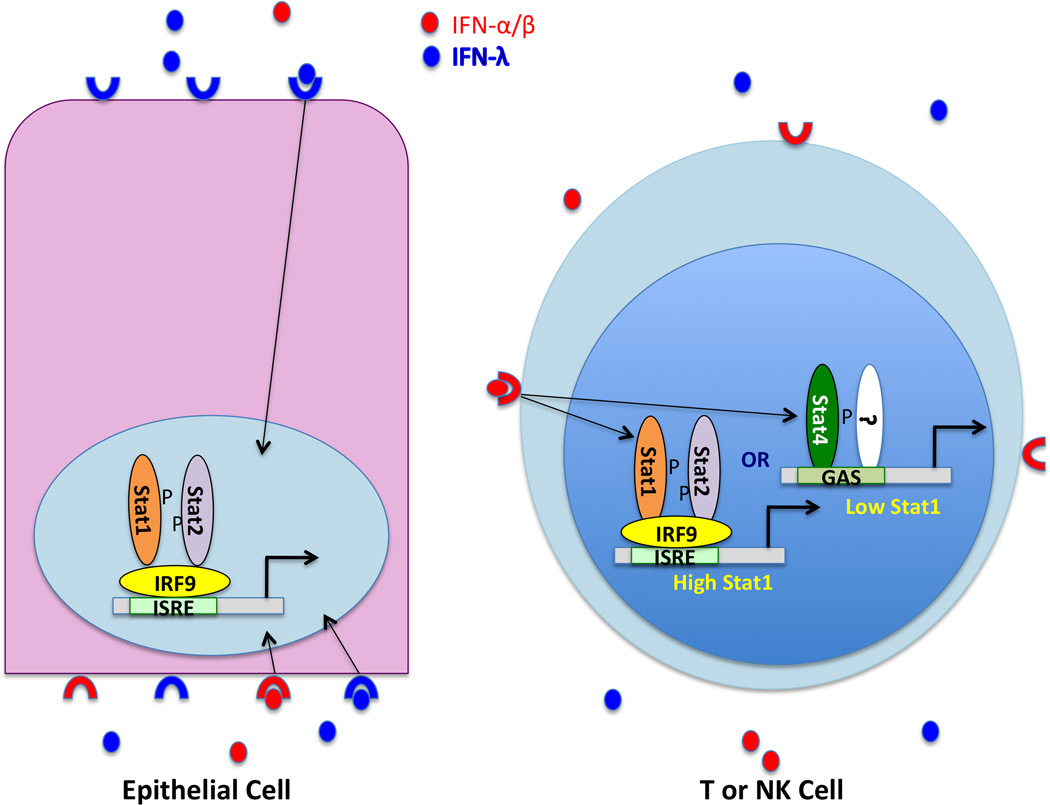
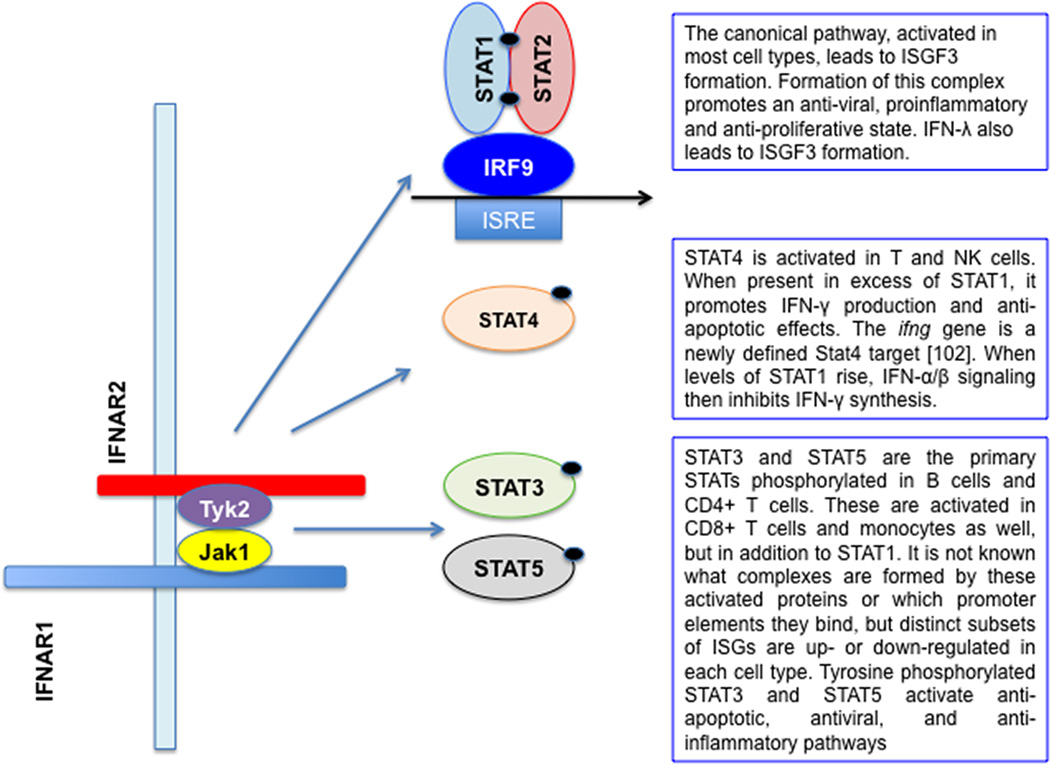
The type I and III interferon (IFN) families consist of cytokines rapidly induced during viral infection that confer antiviral protection on target cells and are critical components of innate immune responses and the transition to effective adaptive immunity. The regulation of their expression involves an intricate and stringently regulated signaling cascade, initiated by recognition most often of viral nucleic acid in cytoplasmic and endosomal compartments and involving a series of protein conformational rearrangements and interactions regulated by helicase action, ubiquitin modification, and protein aggregation, culminating in kinase activation and phosphorylation of critical transcription factors and their regulators. The many IFN subtypes induced by viruses confer amplification, diversification, and cell-type specificity to the host response to infection, providing fertile ground for development of antiviral therapeutics and vaccines.
Figures
References
-
- Iversen MB, Paludan SR. Mechanisms of type III interferon expression. J Interferon Cytokine Res. 2010;30:573–578. - PubMed
-
- Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity. 2006;25:373–381. - PubMed
-
-
Lin WJ, Zheng X, Lin CC, Tsao J, Zhu X, Cody JJ, Coleman JM, Gherzi R, Luo M, Townes TM, et al. Posttranscriptional control of type I interferon genes by KSRP in the innate immune response against viral infection. Mol Cell Biol. 2011;31:3196–3207. Documents a new mechanism for translational control of type I IFN synthesis.
-
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
