

Pathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problems
- PMID: 22327363
- PMCID: PMC3629555
- DOI: 10.1007/s00401-012-0954-z
Pathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problems
Abstract
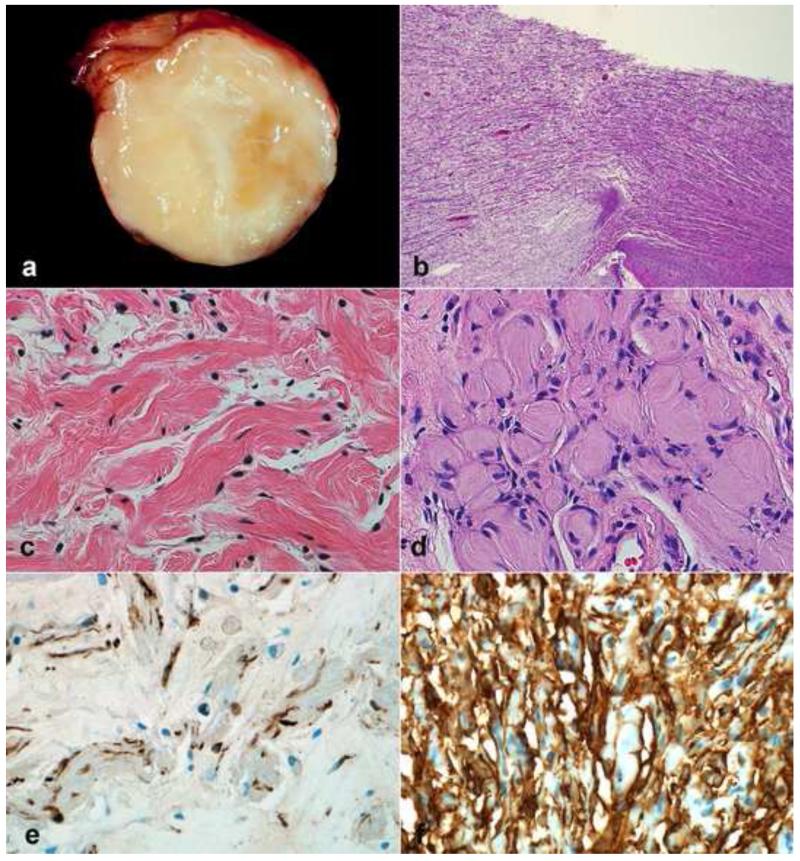
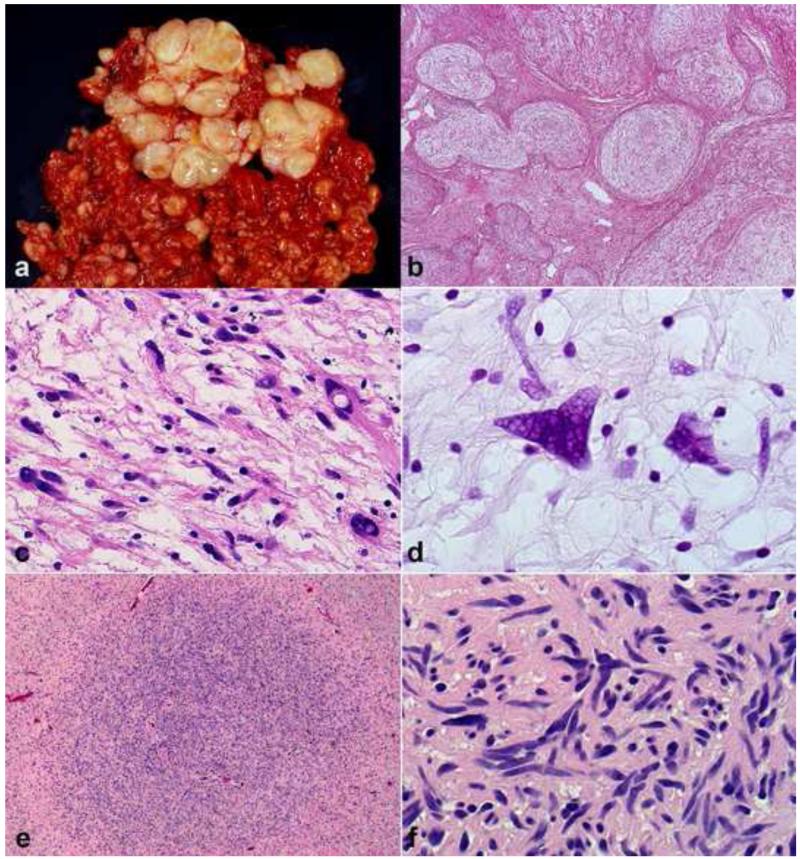
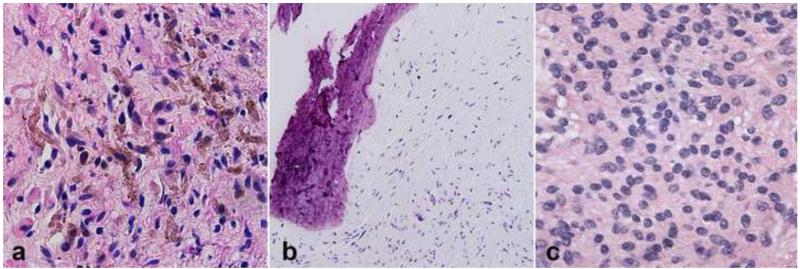
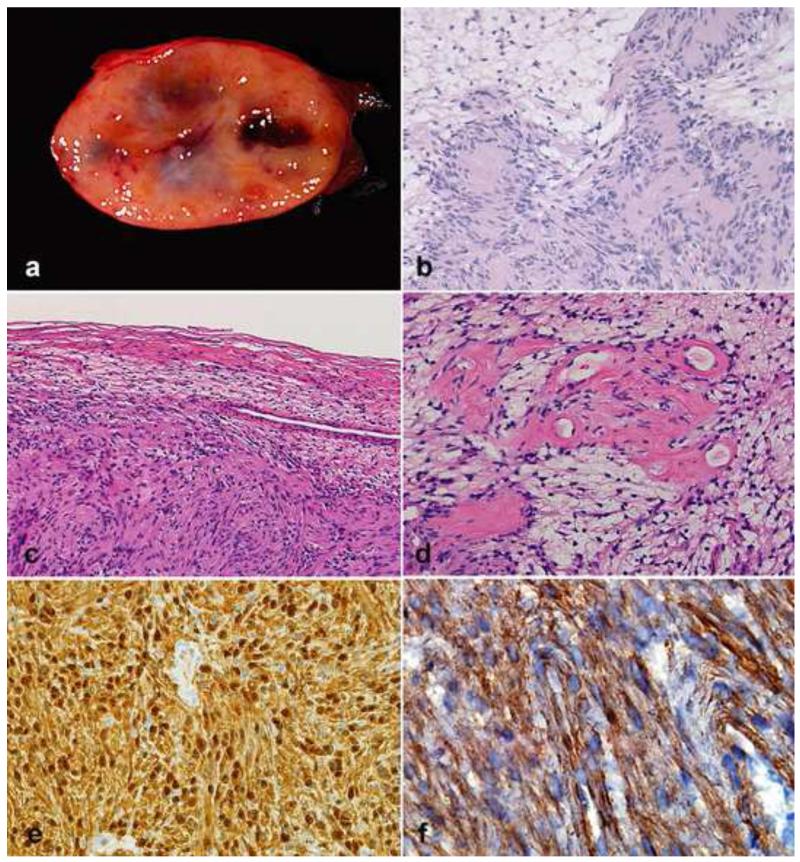
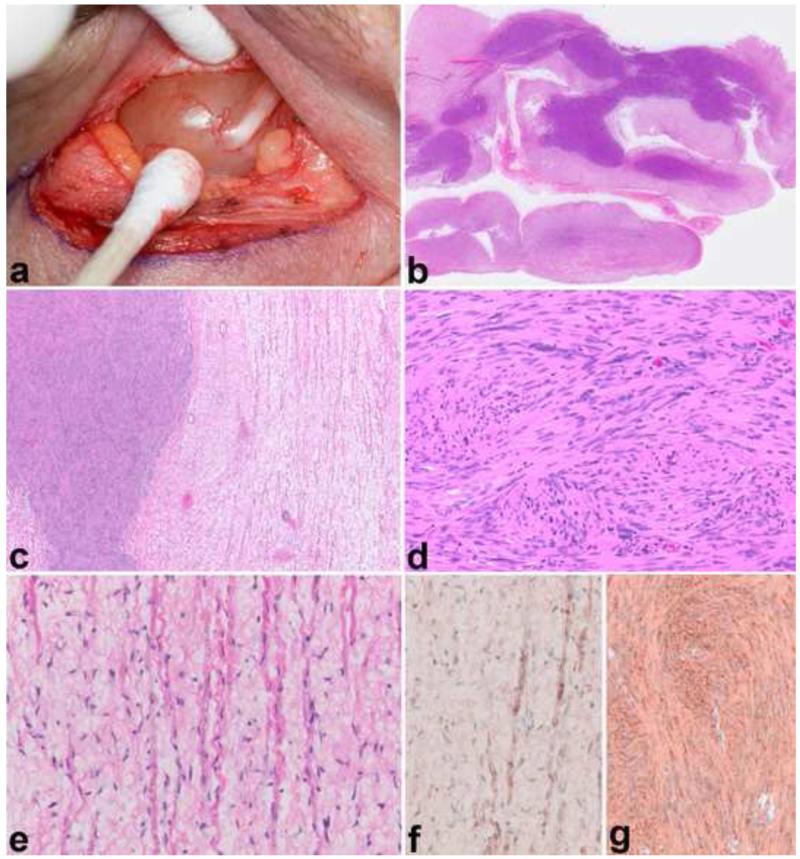
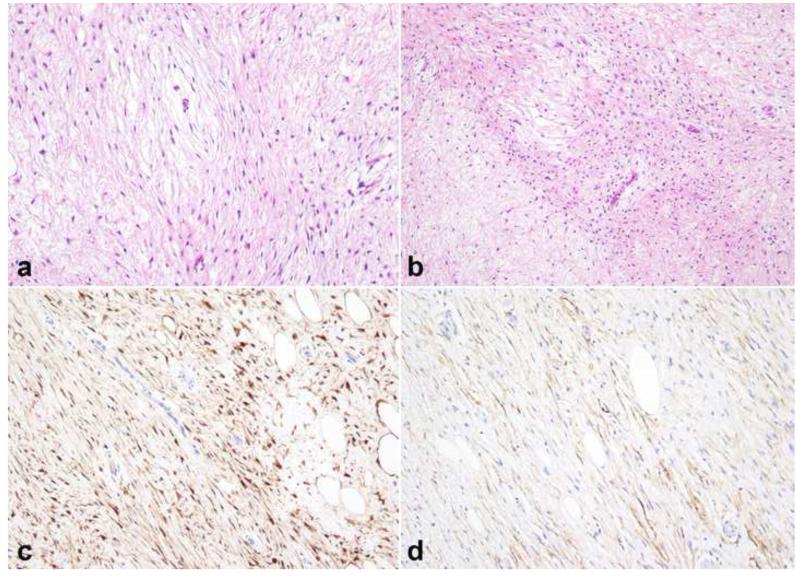
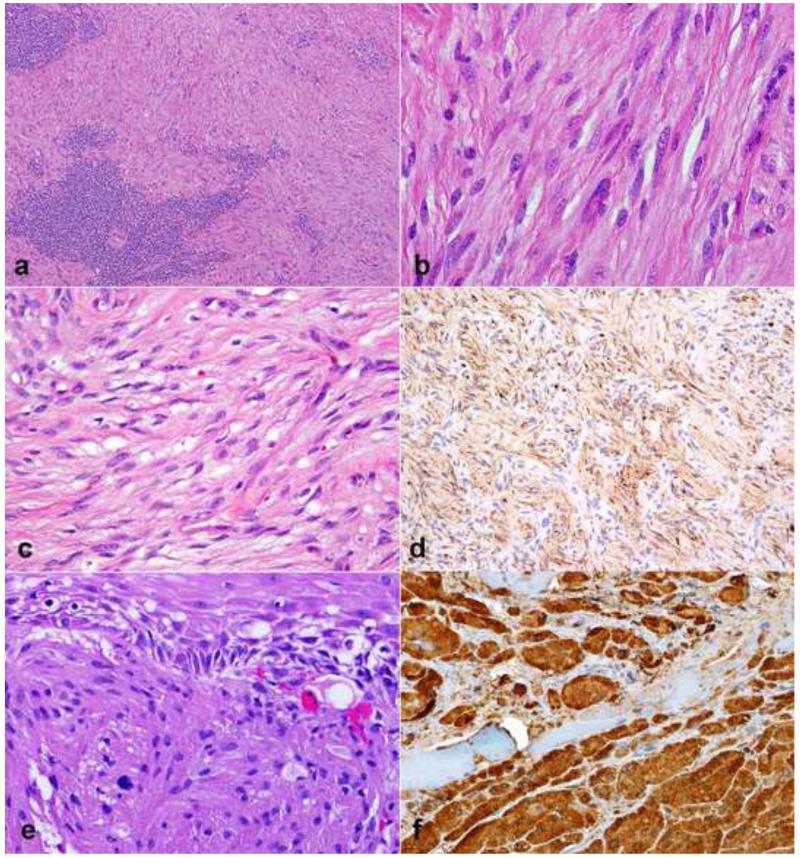
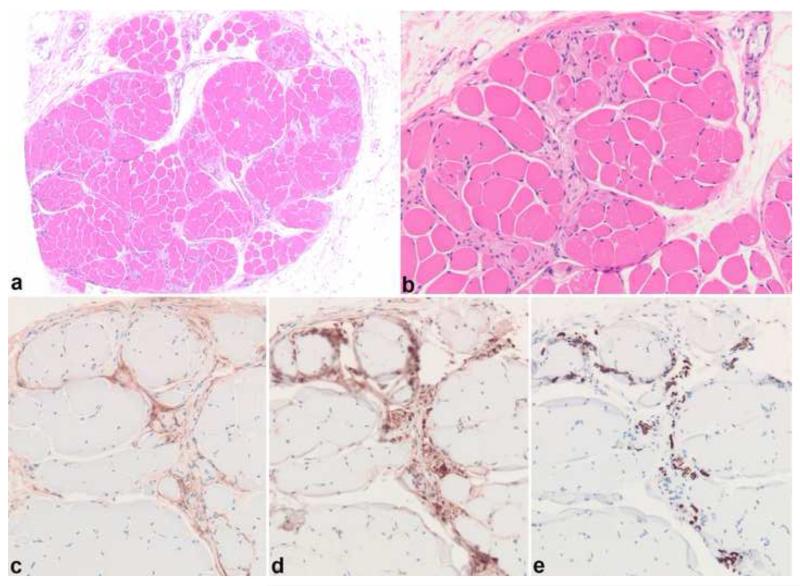
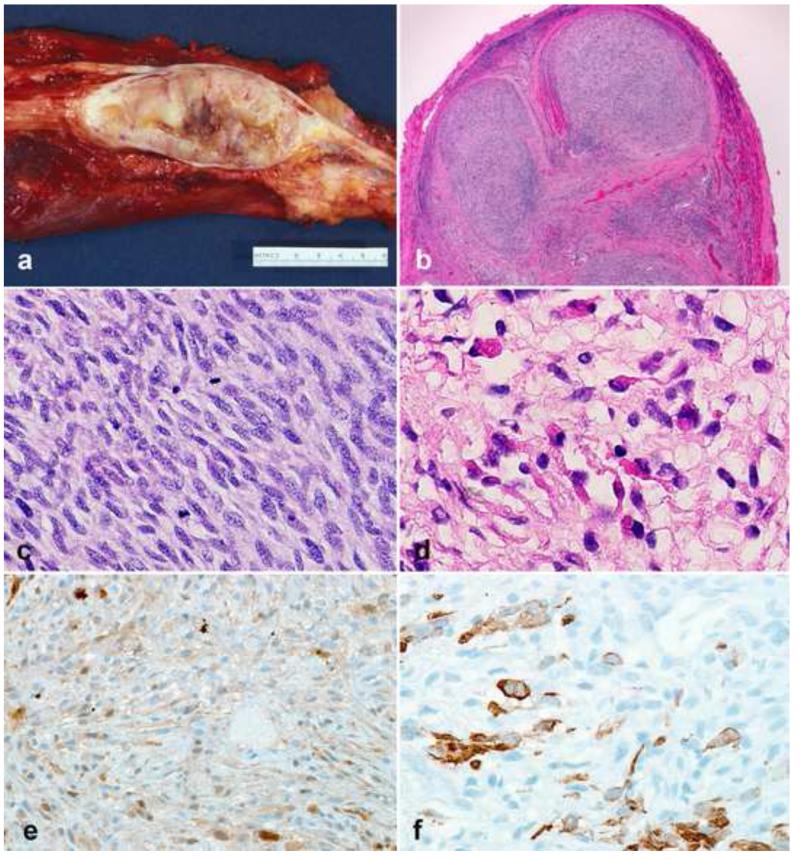
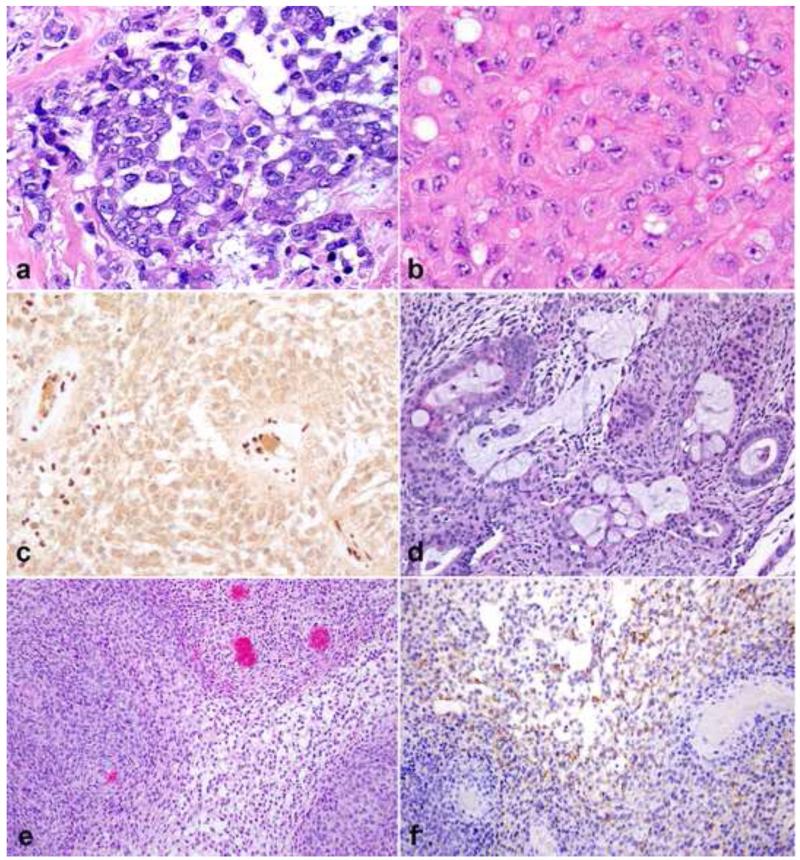
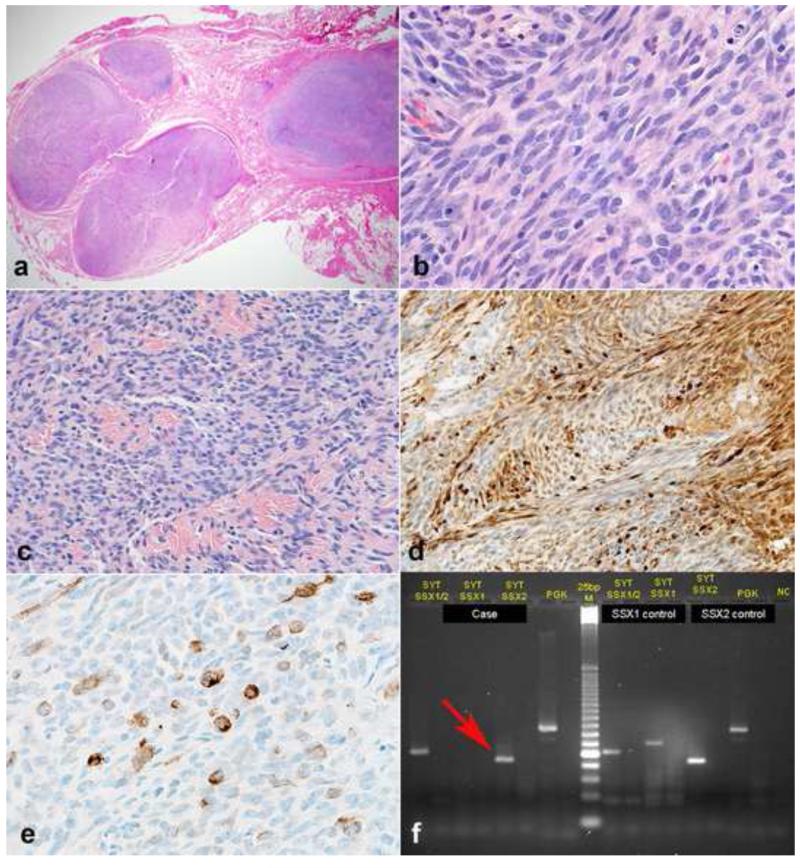
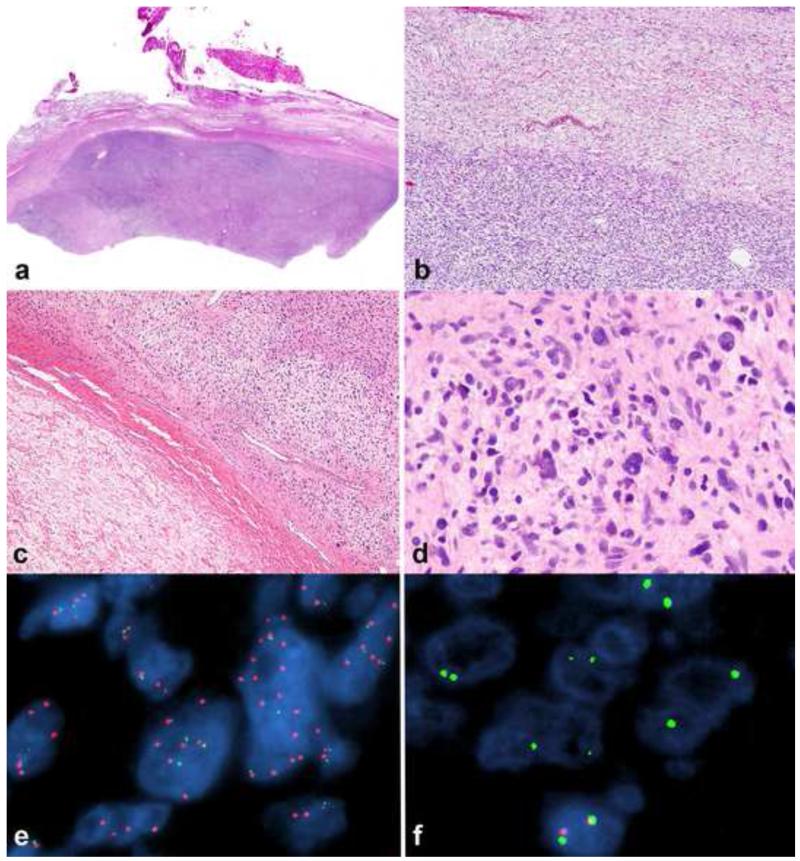
Peripheral nerve sheath tumors are common neoplasms, with classic identifiable features, but on occasion, they are diagnostically challenging. Although well-defined subtypes of peripheral nerve sheath tumors were described early in the history of surgical pathology, controversies regarding the classification and grading of these tumors persist. Advances in molecular biology have provided new insights into the nature of the various peripheral nerve sheath tumors, and have begun to suggest novel targeted therapeutic approaches. In this review, we discuss current concepts and problematic areas in the pathology of peripheral nerve sheath tumors. Diagnostic criteria and differential diagnosis for the major categories of nerve sheath tumors are proposed, including neurofibroma, schwannoma, and perineurioma. Diagnostically challenging variants, including plexiform, cellular and melanotic schwannomas are highlighted. A subset of these affects the childhood population, and has historically been interpreted as malignant, although current evidence and outcome data suggest they represent benign entities. The growing current literature and the author's experience with difficult to classify borderline or "hybrid tumors" are discussed and illustrated. Some of these classification gray zones occur with frequency in the gastrointestinal tract, an anatomical compartment that must always be entertained when examining these neoplasms. Other growing recent areas of interest include the heterogeneous group of pseudoneoplastic lesions involving peripheral nerve composed of mature adipose tissue and/or skeletal muscle, such as the enigmatic neuromuscular choristoma. Malignant peripheral nerve sheath tumors (MPNST) represent a diagnostically controversial group; difficulties in grading and guidelines to separate "atypical neurofibroma" from MPNST are provided. There is an increasing literature of MPNST mimics which neuropathologists must be aware of, including synovial sarcoma and ossifying fibromyxoid tumor. Finally, we discuss entities that are lacking from the section on cranial and paraspinal nerves in the current WHO classification, and that may warrant inclusion in future classifications. In summary, although the diagnosis and classification of most conventional peripheral nerve sheath tumors are relatively straightforward for the experienced observer, yet borderline and difficult-to-classify neoplasms continue to be problematic. In the current review, we attempt to provide some useful guidelines for the surgical neuropathologist to help navigate these persistent, challenging problems.
Figures
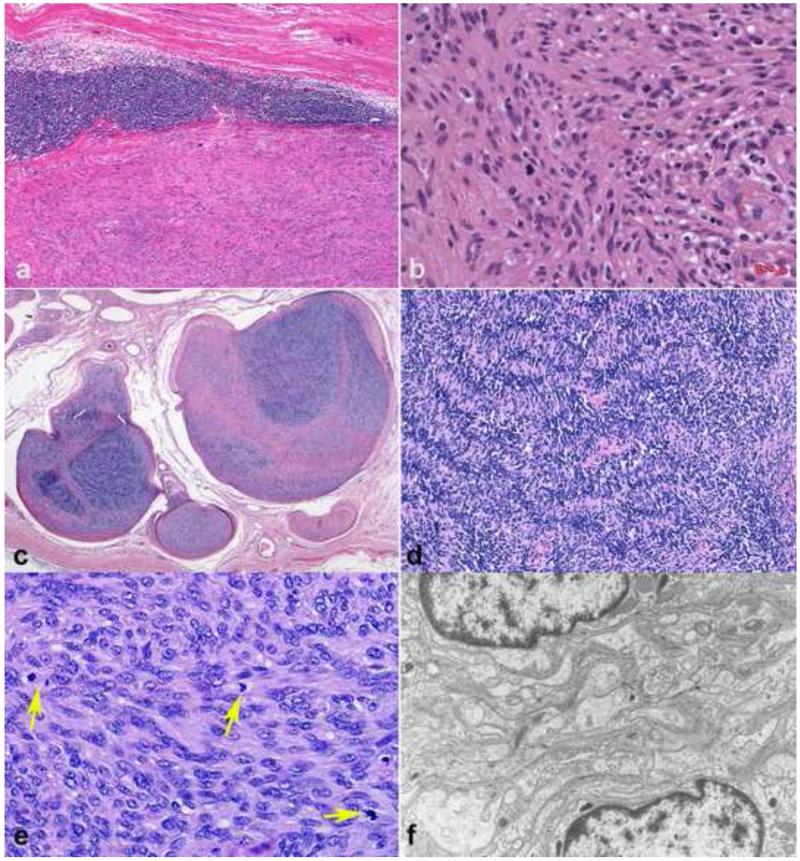
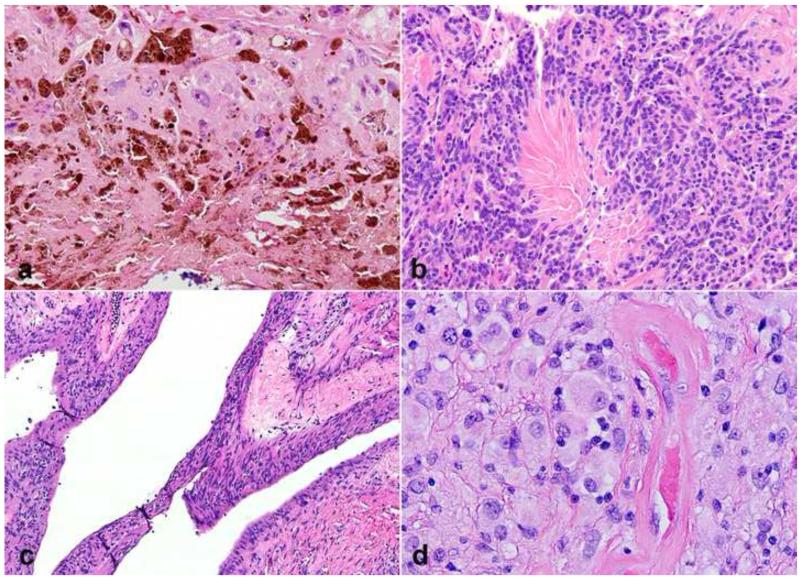
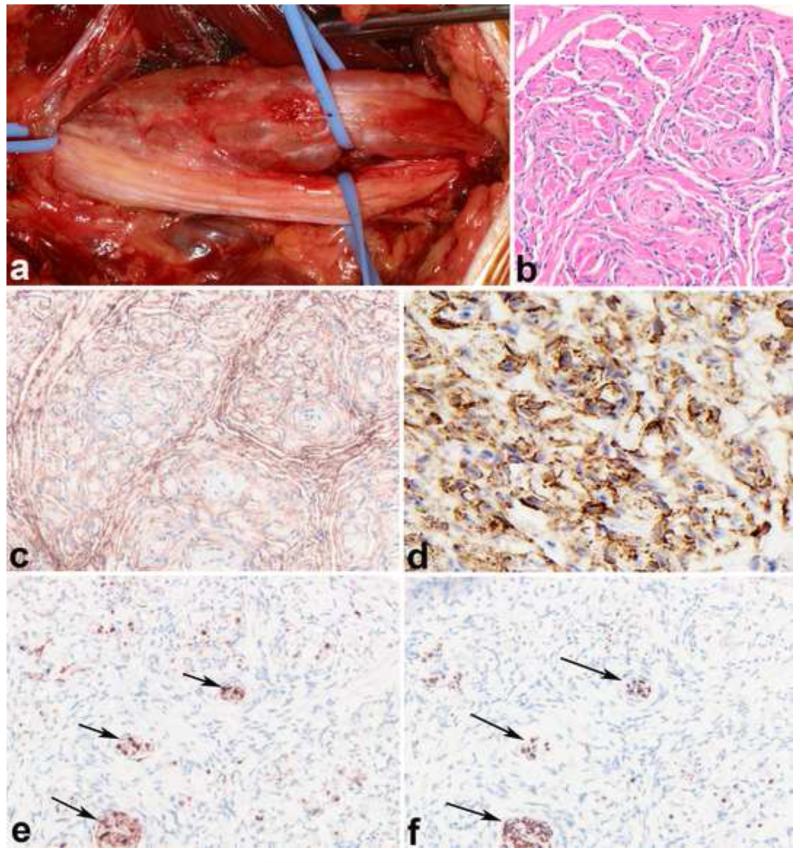
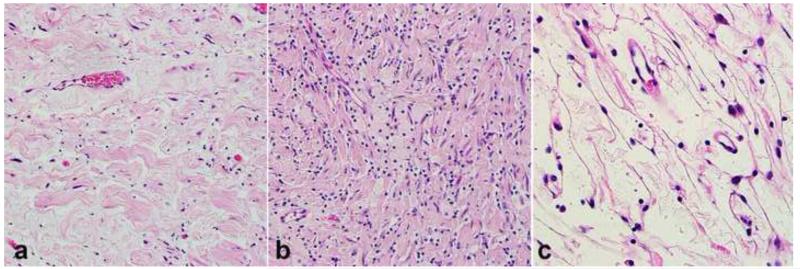
References
-
- Adamiak A, Lee CH, Nielsen TO, Webber D, O’Connell JX. Duodenal epithelioid gastrointestinal stromal tumor with prominent granular cell features. Hum Pathol. 2009;40:599–602. - PubMed
-
- Agaimy A, Wuensch PH. Perineurioma of the stomach. A rare spindle cell neoplasm that should be distinguished from gastrointestinal stromal tumor. Pathol Res Pract. 2005;201:463–467. - PubMed
-
- Agaimy A, Markl B, Kitz J, Wunsch PH, Arnholdt H, Fuzesi L, Hartmann A, Chetty R. Peripheral nerve sheath tumors of the gastrointestinal tract: a multicenter study of 58 patients including NF1-associated gastric schwannoma and unusual morphologic variants. Virchows Arch. 2010;456:411–422. - PubMed
-
- Agaram NP, Prakash S, Antonescu CR. Deep-seated plexiform schwannoma: a pathologic study of 16 cases and comparative analysis with the superficial variety. Am J Surg Pathol. 2005;29:1042–1048. - PubMed
-
- Ahrens WA, Ridenour RV, 3rd, Caron BL, Miller DV, Folpe AL. GLUT-1 expression in mesenchymal tumors: an immunohistochemical study of 247 soft tissue and bone neoplasms. Hum Pathol. 2008;39:1519–1526. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
