

NOX4 pathway as a source of selective insulin resistance and responsiveness
- PMID: 22328777
- PMCID: PMC3398464
- DOI: 10.1161/ATVBAHA.111.244525
NOX4 pathway as a source of selective insulin resistance and responsiveness
Abstract
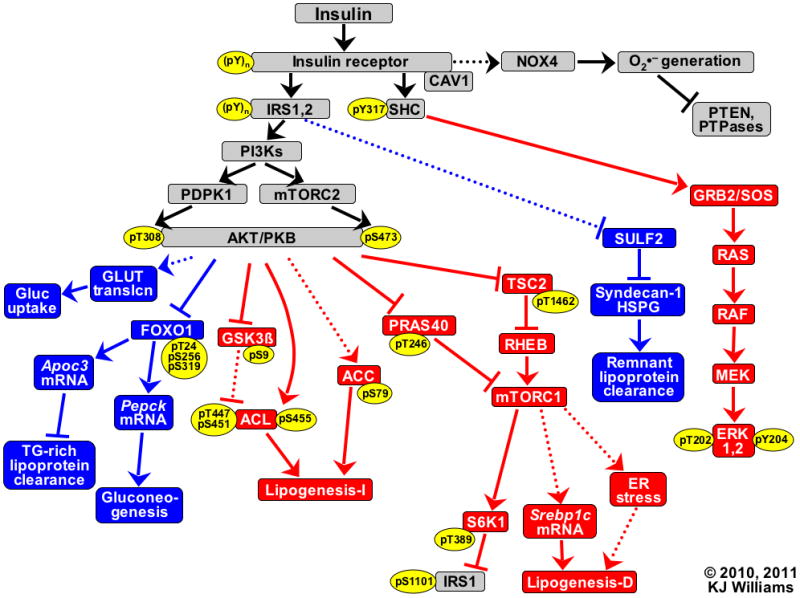
Objective: Type 2 diabetes mellitus and related syndromes exhibit a deadly triad of dyslipoproteinemia, which leads to atherosclerosis; hyperglycemia, which causes microvascular disease; and hypertension. These features share a common, but unexplained, origin-namely, pathway-selective insulin resistance and responsiveness. Here, we undertook a comprehensive characterization of pathway-selective insulin resistance and responsiveness in liver and hepatocytes by examining 18 downstream targets of the insulin receptor, surveying the AKT, ERK, and NAD(P)H oxidase 4 pathways.
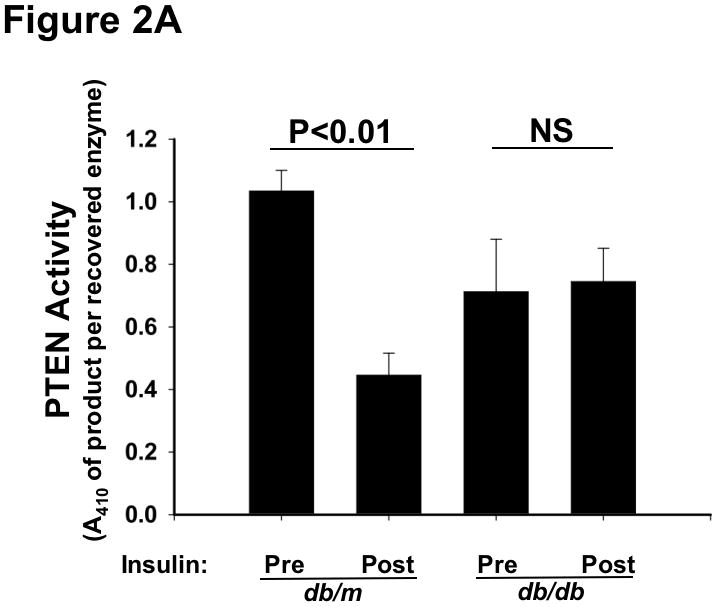
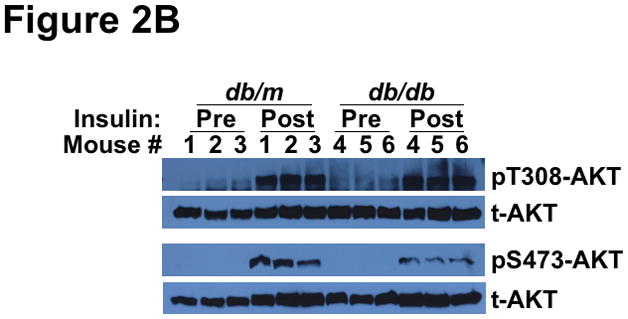
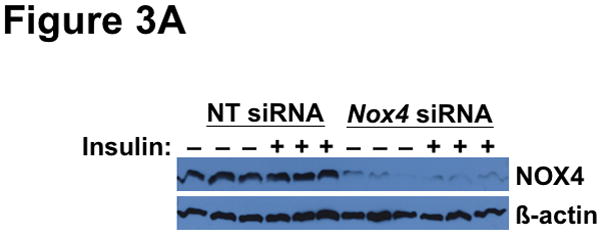
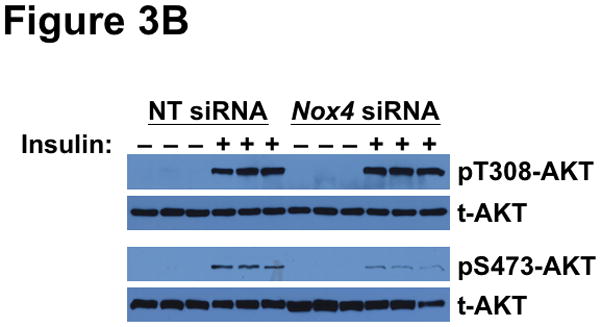
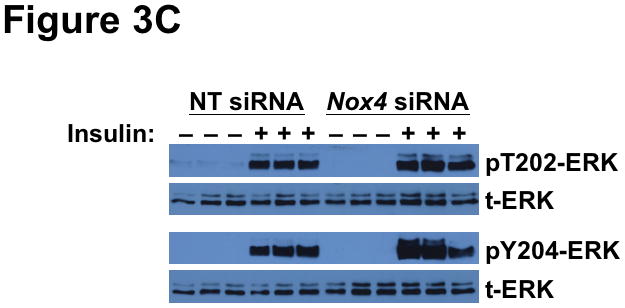
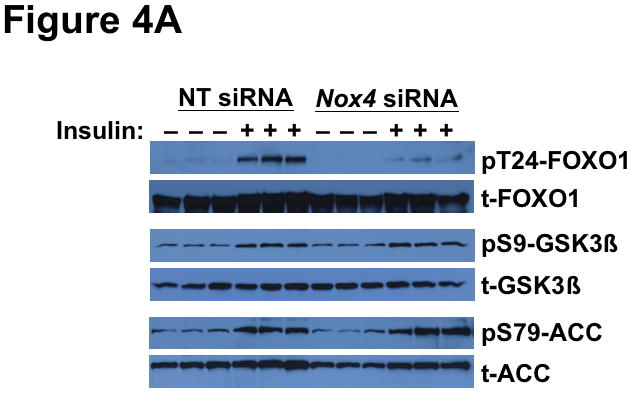
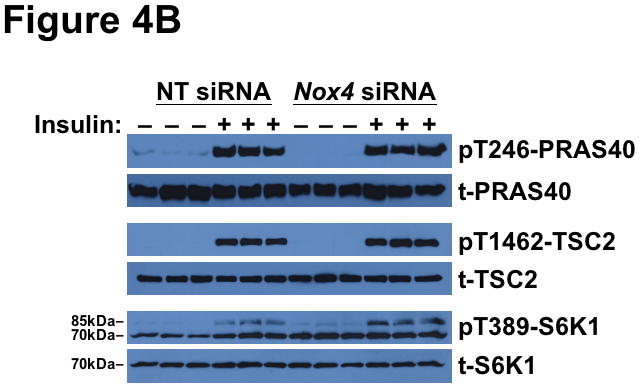
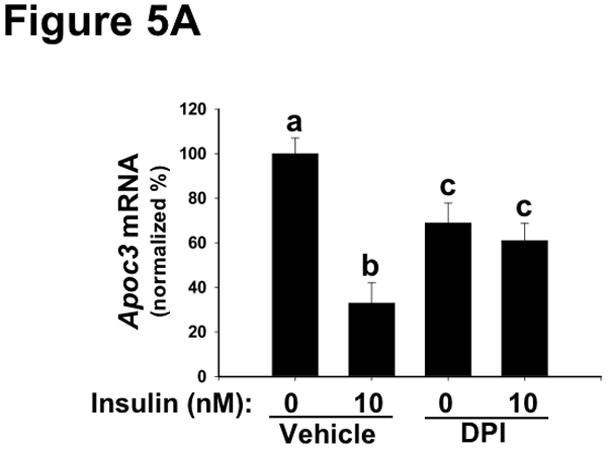
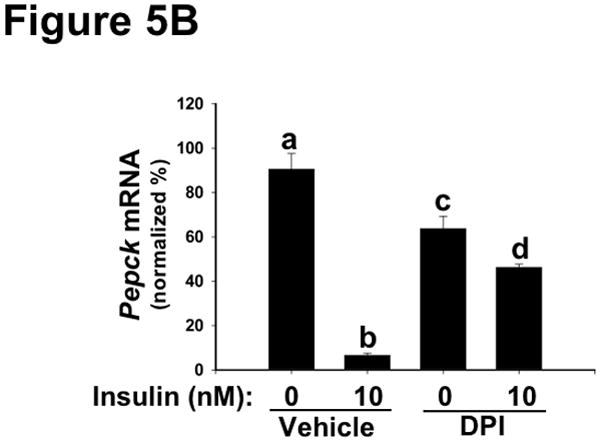
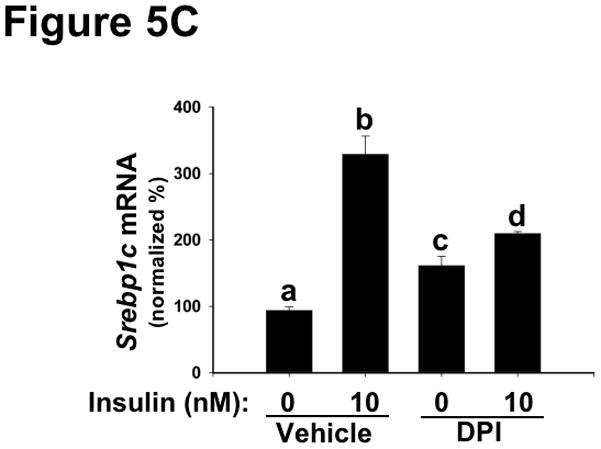
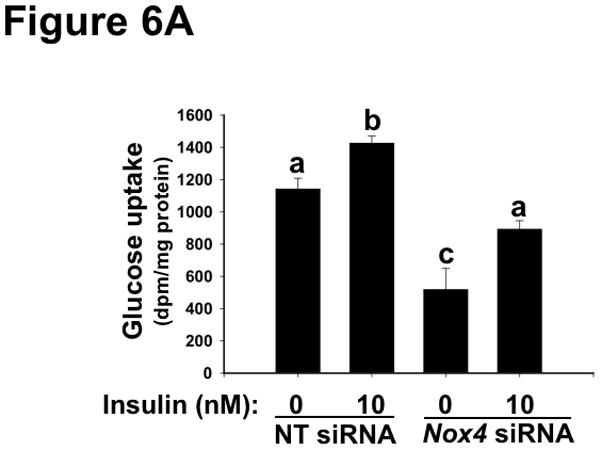
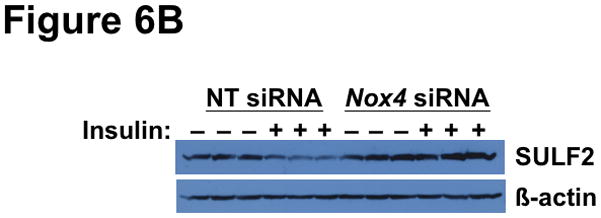
Methods and results: Injection of insulin into hyperphagic, obese type 2 diabetic db/db mice failed to inactivate hepatic protein tyrosine phosphatase gene family members, a crucial action of NAD(P)H oxidase 4 previously thought to be required for all signaling through AKT and ERK. Insulin-stimulated type 2 diabetic livers unexpectedly produced an unusual form of AKT that was phosphorylated at Thr308 (pT308), with only weak insulin-stimulated phosphorylation at Ser473. Remarkably, knockdown or inhibition of NAD(P)H oxidase 4 in cultured hepatocytes recapitulated the entire complicated pattern of pathway-selective insulin resistance and responsiveness seen in vivo-namely, monophosphorylated pT308-AKT, impaired insulin-stimulated pathways for lowering plasma lipids and glucose, but continued lipogenic pathways and robust ERK activation.
Conclusions: Functional disturbance of a single molecule, NAD(P)H oxidase 4, is sufficient to induce the key harmful features of deranged insulin signaling in type 2 diabetes mellitus, obesity, and other conditions associated with hyperinsulinemia and pathway-selective insulin resistance and responsiveness.
Conflict of interest statement
No conflicts of interest to report (XW, KJW).
Figures
Comment in
-
kNOXing on the door of selective insulin resistance.Arterioscler Thromb Vasc Biol. 2012 May;32(5):1063-5. doi: 10.1161/ATVBAHA.112.246868. Arterioscler Thromb Vasc Biol. 2012. PMID: 22517360 Free PMC article. No abstract available.
References
-
- Shimomura I, Matsuda M, Hammer RE, Bashmakov Y, Brown MS, Goldstein JL. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell. 2000;6:77–86. - PubMed
-
- Muniyappa R, Montagnani M, Koh KK, Quon MJ. Cardiovascular actions of insulin. Endocr Rev. 2007;28:463–491. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
