

Androgen-induced activation of gonadotropin-regulated testicular RNA helicase (GRTH/Ddx25) transcription: essential role of a nonclassical androgen response element half-site
- PMID: 22331472
- PMCID: PMC3318589
- DOI: 10.1128/MCB.06002-11
Androgen-induced activation of gonadotropin-regulated testicular RNA helicase (GRTH/Ddx25) transcription: essential role of a nonclassical androgen response element half-site
Abstract
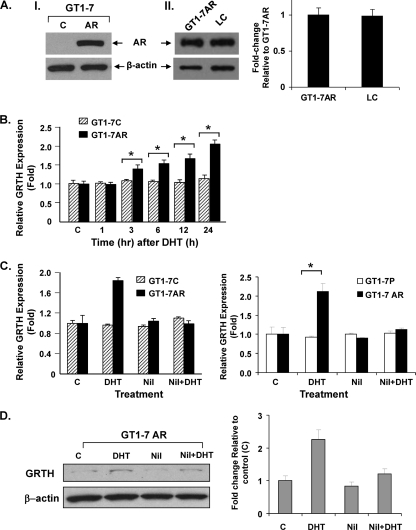
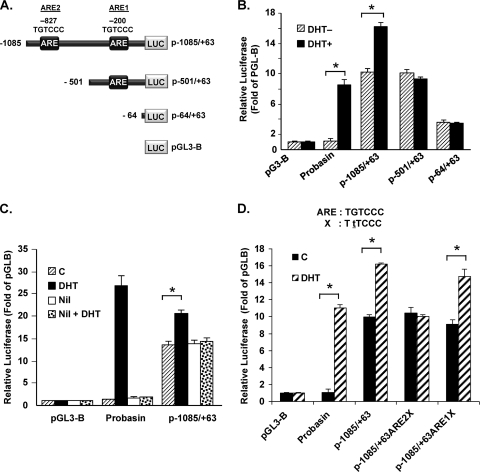
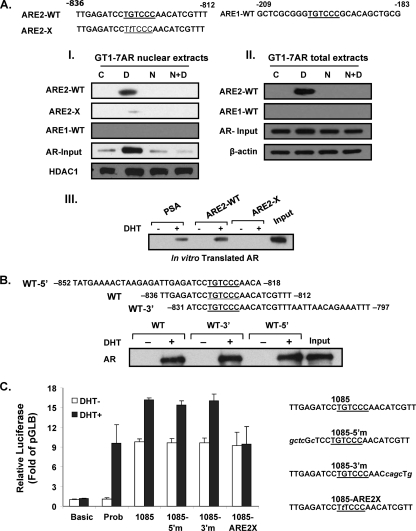
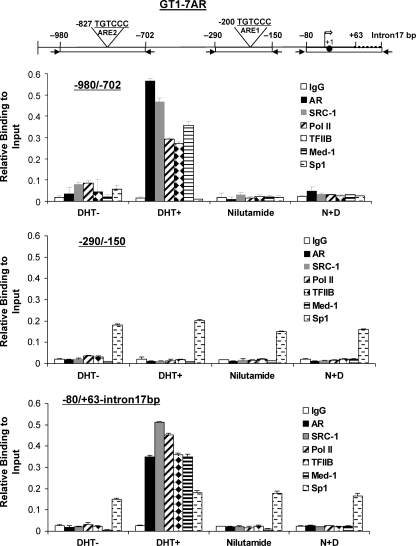
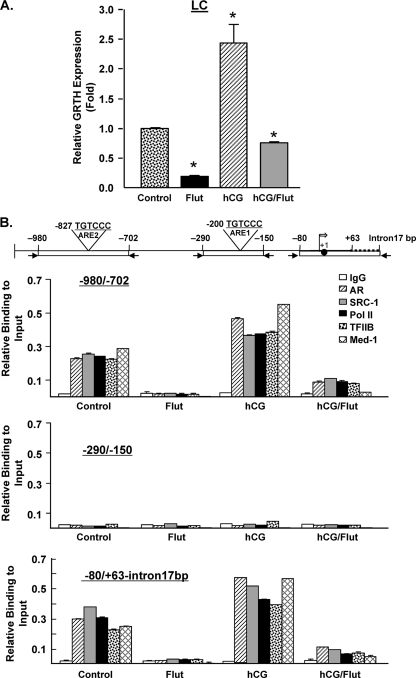
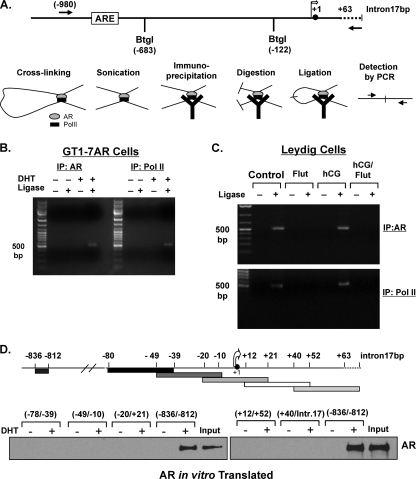
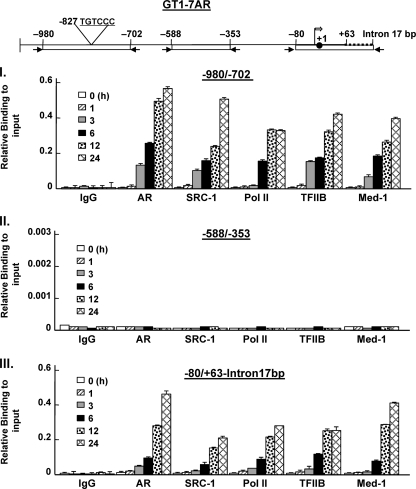
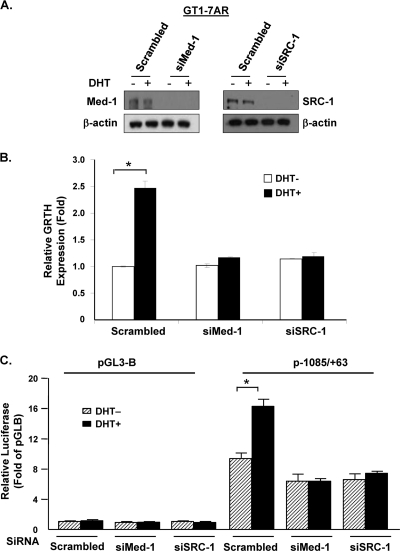
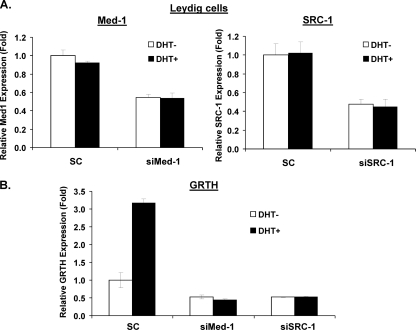
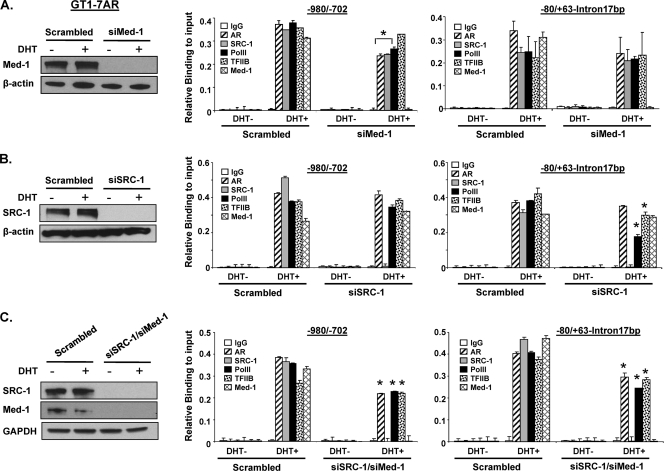
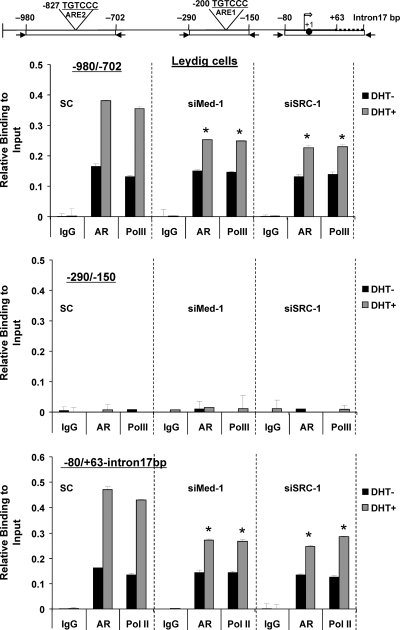
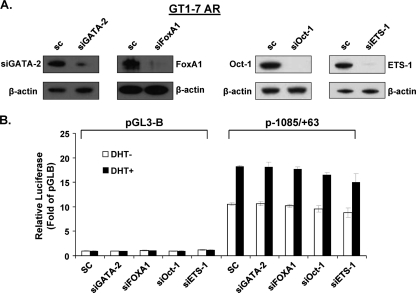
GRTH, a testis-specific member of the DEAD-box family of RNA helicases essential for spermatogenesis, is present in Leydig cells (LC) and germ cells. In LC, it exerts an autocrine negative regulation on androgen production induced by gonadotropin. GRTH is transcriptionally upregulated by gonadotropin via cyclic AMP/androgen through androgen receptors (AR). For studies of GRTH regulation by androgen in LC, we utilized in vitro/in vivo models. Androgen-induced GRTH expression was prevented by an AR antagonist. Two putative atypical ARE half-sites are present at bp -200 and -827 (ARE1 and ARE2). Point mutation of ARE2 prevented androgen-induced AR binding/function and upregulation of GRTH transcription. Chromatin immunoprecipitation (ChIP) assays showed recruitment of AR, SRC-1, Med-1, transcription factor IIB (TFIIB), and polymerase II (PolII) to GRTH ARE2 (bp -980/-702) and to the promoter region (bp -80/+63). ChIP3C assays revealed short-range chromosomal looping between AR/ARE2 and the core transcriptional machinery at the promoter. Knockdown of Med-1 and/or SRC-1 demonstrated the presence of a nonproductive complex which included AR, TFIIB, and PolII and the essential role of these coactivators in the transcriptional activation of GRTH. Our findings provide new insights into the molecular mechanism of androgen-regulated transcription in LC.
Figures

References
-
- Belakavadi M, Fondell JD. 2006. Role of the mediator complex in nuclear hormone receptor signaling. Rev. Physiol. Biochem. Pharmacol. 156: 23–43 - PubMed
-
- Bennett NC, Gardiner RA, Hooper JD, Johnson DW, Gobe GC. 2010. Molecular cell biology of androgen receptor signalling. Int. J. Biochem. Cell Biol. 42: 813–827 - PubMed
-
- Briegel K, et al. 1993. Ectopic expression of a conditional GATA-2/estrogen receptor chimera arrests erythroid differentiation in a hormone-dependent manner. Genes Dev. 7: 1097–1109 - PubMed
-
- Collins LL, et al. 2003. The androgen receptor in spermatogenesis. Cytogenet. Genome Res. 103: 299–301 - PubMed
-
- Dekker J, Rippe K, Dekker M, Kleckner N. 2002. Capturing chromosome conformation. Science 295: 1306–1311 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous