

Ganglioside GM1 induces phosphorylation of mutant huntingtin and restores normal motor behavior in Huntington disease mice
- PMID: 22331905
- PMCID: PMC3295265
- DOI: 10.1073/pnas.1114502109
Ganglioside GM1 induces phosphorylation of mutant huntingtin and restores normal motor behavior in Huntington disease mice
Abstract
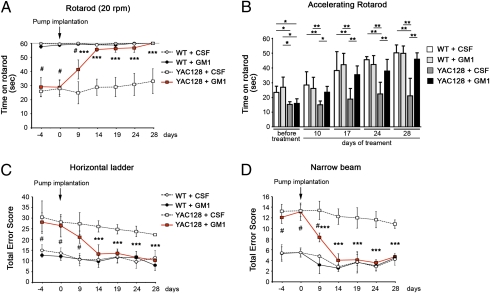
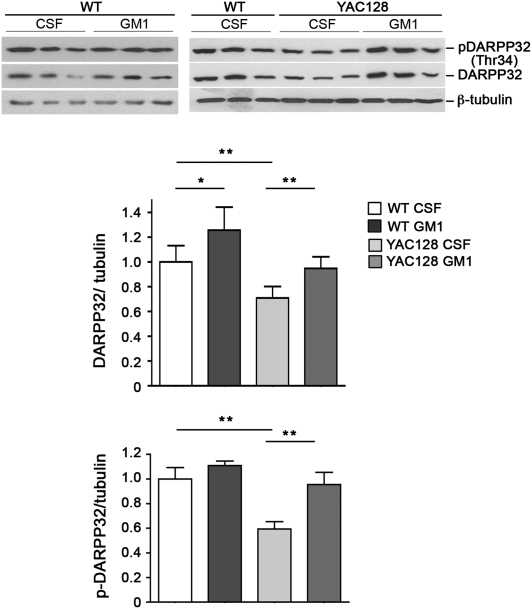
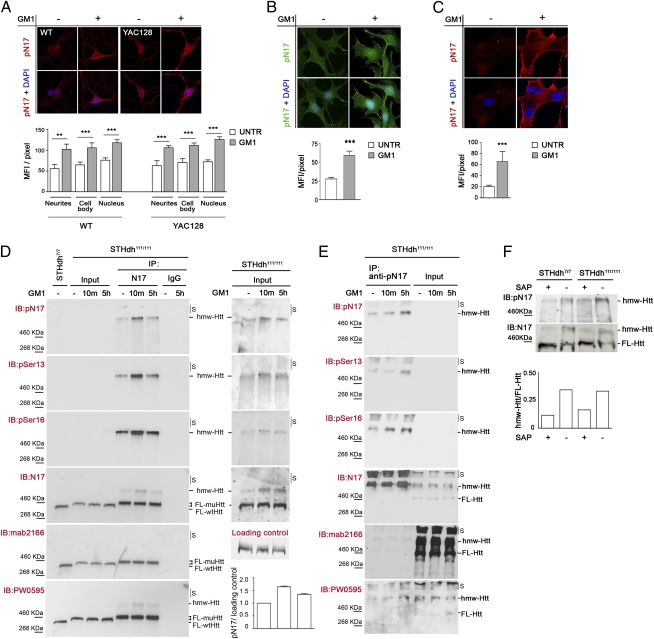
Huntington disease (HD) is a progressive neurodegenerative monogenic disorder caused by expansion of a polyglutamine stretch in the huntingtin (Htt) protein. Mutant huntingtin triggers neural dysfunction and death, mainly in the corpus striatum and cerebral cortex, resulting in pathognomonic motor symptoms, as well as cognitive and psychiatric decline. Currently, there is no effective treatment for HD. We report that intraventricular infusion of ganglioside GM1 induces phosphorylation of mutant huntingtin at specific serine amino acid residues that attenuate huntingtin toxicity, and restores normal motor function in already symptomatic HD mice. Thus, our studies have identified a potential therapy for HD that targets a posttranslational modification of mutant huntingtin with critical effects on disease pathogenesis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Zuccato C, Valenza M, Cattaneo E. Molecular mechanisms and potential therapeutical targets in Huntington's disease. Physiol Rev. 2010;90:905–981. - PubMed
-
- Steffan JS, et al. SUMO modification of Huntingtin and Huntington's disease pathology. Science. 2004;304:100–104. - PubMed
-
- Graham RK, et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. - PubMed
-
- Schilling B, et al. Huntingtin phosphorylation sites mapped by mass spectrometry. Modulation of cleavage and toxicity. J Biol Chem. 2006;281:23686–23697. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
