

Positional correlation analysis improves reconstruction of full-length transcripts and alternative isoforms from noisy array signals or short reads
- PMID: 22332235
- PMCID: PMC3315713
- DOI: 10.1093/bioinformatics/bts065
Positional correlation analysis improves reconstruction of full-length transcripts and alternative isoforms from noisy array signals or short reads
Abstract
Motivation: A reconstruction of full-length transcripts observed by next-generation sequencer or tiling arrays is an essential technique to know all phenomena of transcriptomes. Several techniques of the reconstruction have been developed. However, problems of high-level noises and biases still remain and interrupt the reconstruction. A method is required that is robust against noise and bias and correctly reconstructs transcripts regardless of equipment used.
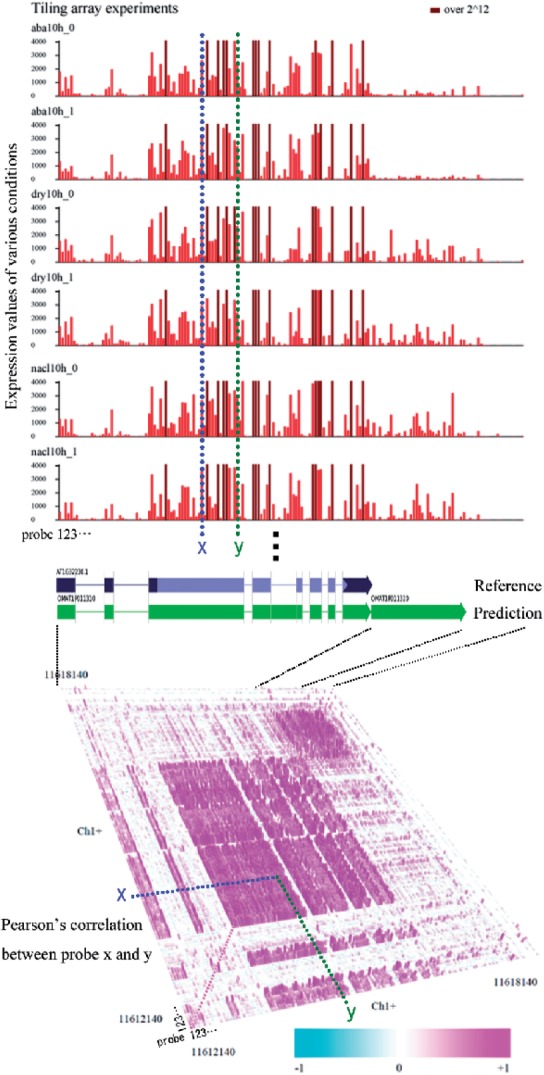
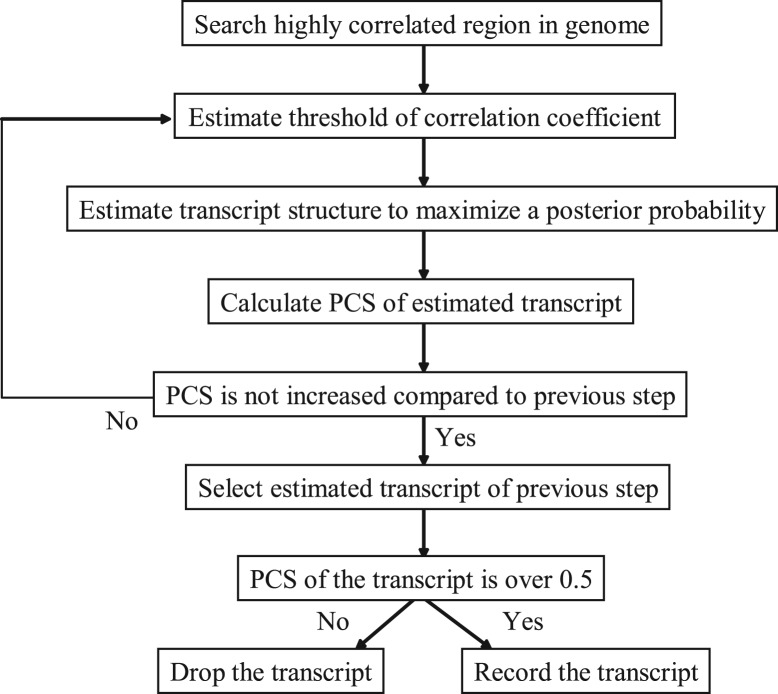
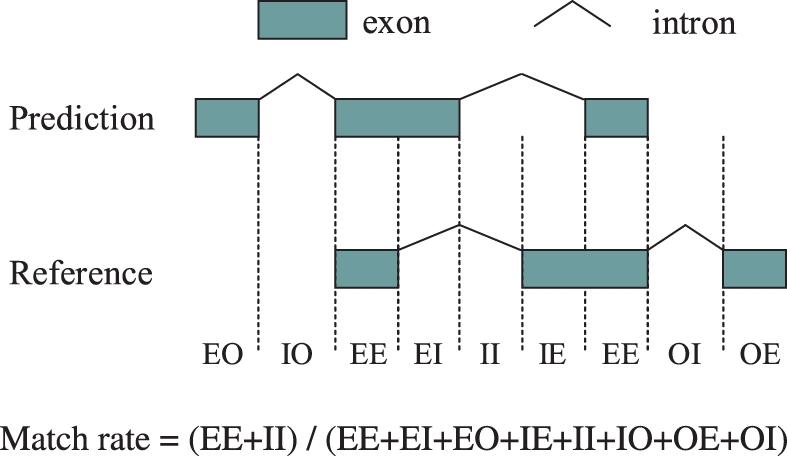
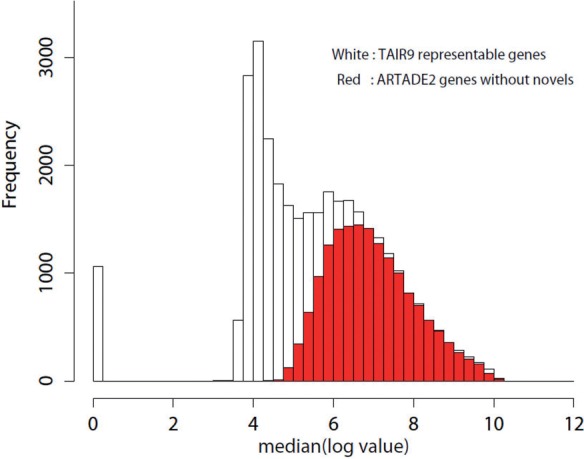
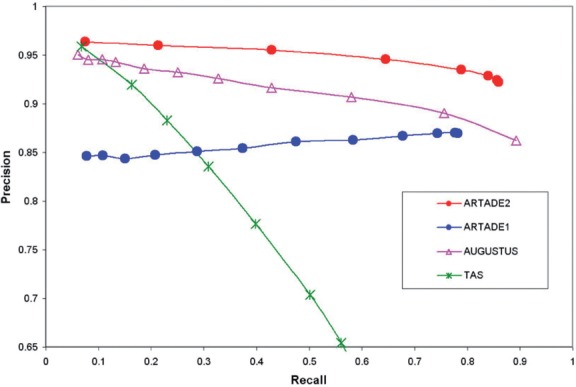
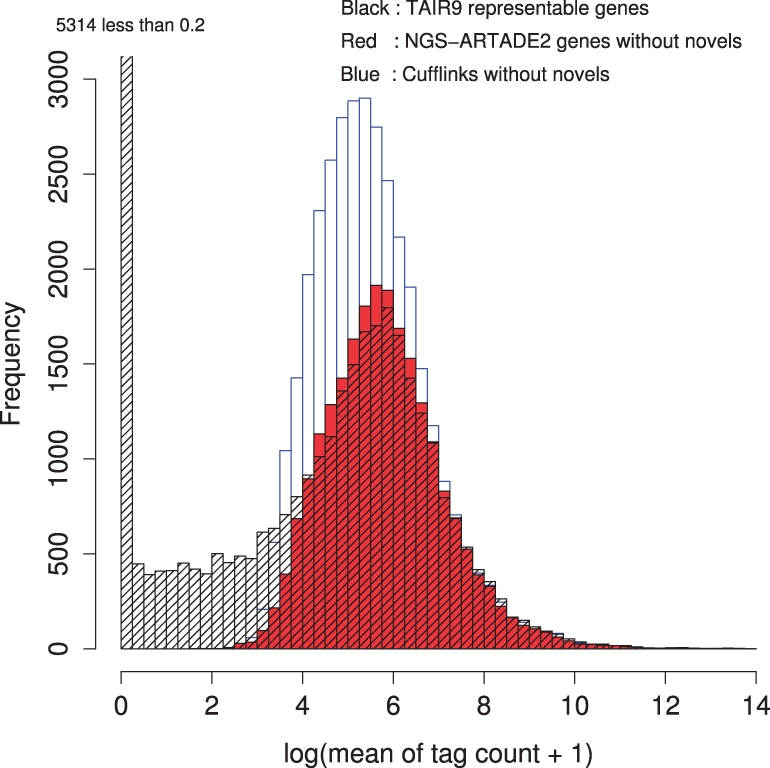
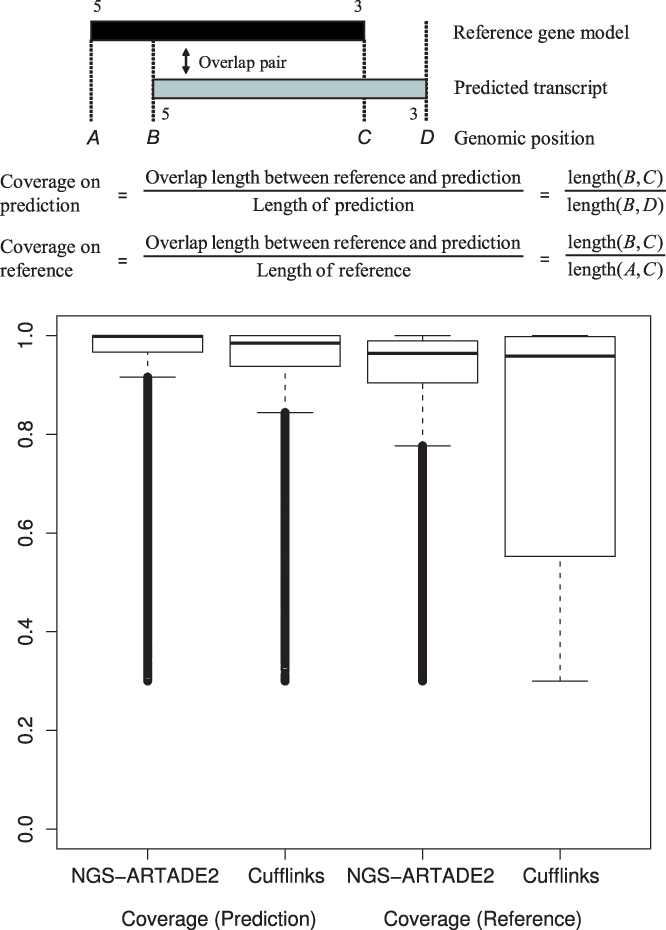
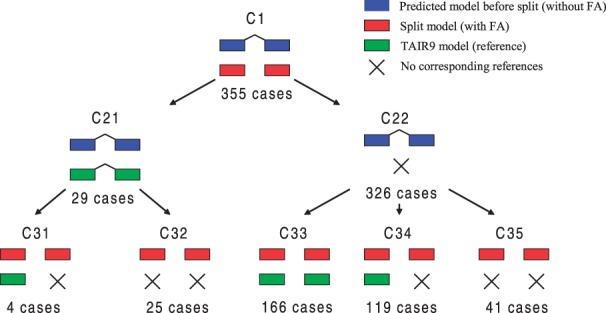
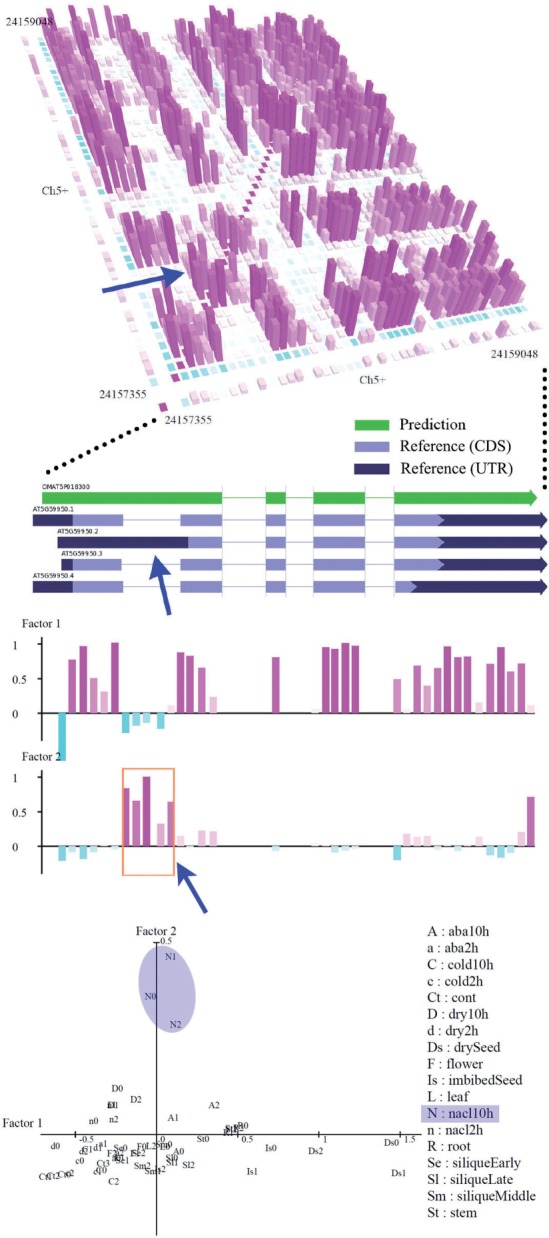
Results: We propose a completely new statistical method that reconstructs full-length transcripts and can be applied on both next-generation sequencers and tiling arrays. The method called ARTADE2 analyzes 'positional correlation', meaning correlations of expression values for every combination on genomic positions of multiple transcriptional data. ARTADE2 then reconstructs full-length transcripts using a logistic model based on the positional correlation and the Markov model. ARTADE2 elucidated 17 591 full-length transcripts from 55 transcriptome datasets and showed notable performance compared with other recent prediction methods. Moreover, 1489 novel transcripts were discovered. We experimentally tested 16 novel transcripts, among which 14 were confirmed by reverse transcription-polymerase chain reaction and sequence mapping. The method also showed notable performance for reconstructing of mRNA observed by a next-generation sequencer. Moreover, the positional correlation and factor analysis embedded in ARTADE2 successfully detected regions at which alternative isoforms may exist, and thus are expected to be applied for discovering transcript biomarkers for a wide range of disciplines including preemptive medicine.
Availability: http://matome.base.riken.jp
Contact: toyoda@base.riken.jp
Supplementary information: Supplementary data are available at Bioinformatics online.
Figures

References
-
- Baerenfaller K., et al. Genome-scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science. 2008;320:938–941. - PubMed
-
- German M.A., et al. Construction of Parallel Analysis of RNA Ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. Nat. Protoc. 2009;4:356–362. - PubMed
-
- Hendrickson A.E., White P.O. PROMAX : a quick method for rotation to oblique simple structure. Br. J. Stat. Psychol. 1964;17:65–70.
