

Combined spectroscopic/computational studies of vitamin B12 precursors: geometric and electronic structures of cobinamides
- PMID: 22332807
- PMCID: PMC4656031
- DOI: 10.1021/ic202052g
Combined spectroscopic/computational studies of vitamin B12 precursors: geometric and electronic structures of cobinamides
Abstract
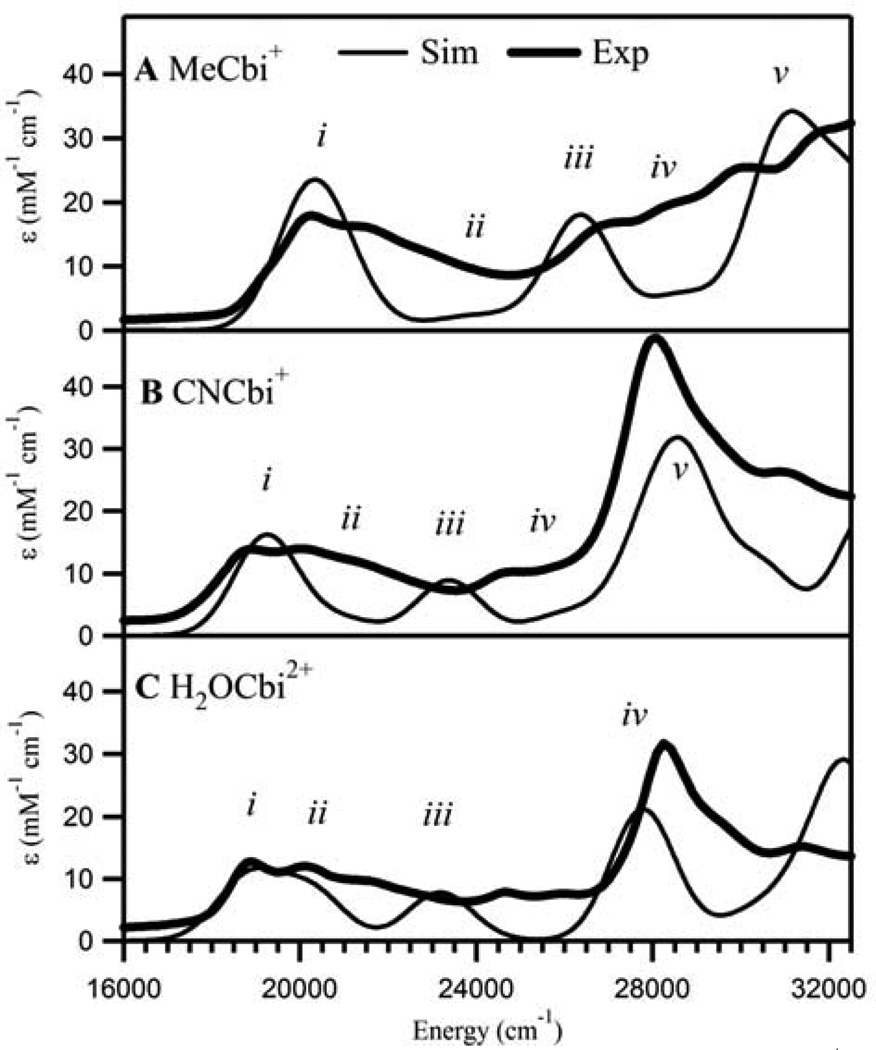
Vitamin B(12) (cyanocobalamin) and its biologically active derivatives, methylcobalamin and adenosylcobalamin, are members of the family of corrinoids, which also includes cobinamides. As biological precursors to cobalamins, cobinamides possess the same structural core, consisting of a low-spin Co(3+) ion that is ligated equatorially by the four nitrogens of a highly substituted tetrapyrrole macrocycle (the corrin ring), but differ with respect to the lower axial ligation. Specifically, cobinamides possess a water molecule instead of the nucleotide loop that coordinates axially to Co(3+)cobalamins via its dimethylbenzimidazole (DMB) base. Compared to the cobalamin species, cobinamides have proven much more difficult to study experimentally, thus far eluding characterization by X-ray crystallography. In this study, we have utilized combined quantum mechanics/molecular mechanics (QM/MM) computations to generate complete structural models of a representative set of cobinamide species with varying upper axial ligands. To validate the use of this approach, analogous QM/MM geometry optimizations were carried out on entire models of the cobalamin counterparts for which high-resolution X-ray structural data are available. The accuracy of the cobinamide structures was assessed further by comparing electronic absorption spectra computed using time-dependent density functional theory to those obtained experimentally. Collectively, the results obtained in this study indicate that the DMB → H(2)O lower axial ligand switch primarily affects the energies of the Co 3d(z(2))-based molecular orbital (MO) and, to a lesser extent, the other Co 3d-based MOs as well as the corrin π-based highest energy MO. Thus, while the energy of the lowest-energy electronic transition of cobalamins changes considerably as a function of the upper axial ligand, it is nearly invariant for the cobinamides.
Figures









References
-
- Comba P, Remenyi R. Coord. Chem. Rev. 2003;238–239:9–20.
-
- Deeth RJ. Comprehensive Coordination Chemistry II. 2004;2:457–465.
-
- Christianson DW, Lipscomb WN. J. Am. Chem. Soc. 1985;107:2682–2686.
-
- Jensen KP, Ryde U. J. Phys. Chem. A. 2003;107:7539–7545.
-
- Noodleman L, Lovell T, Han W-G, Liu T, Torres RA, Himo F. Comprehensive Coordination Chemistry II. 2004;2:491–510.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
