

A pRNA-induced structural rearrangement triggers 6S-1 RNA release from RNA polymerase in Bacillus subtilis
- PMID: 22333917
- PMCID: PMC3321203
- DOI: 10.1038/emboj.2012.23
A pRNA-induced structural rearrangement triggers 6S-1 RNA release from RNA polymerase in Bacillus subtilis
Abstract
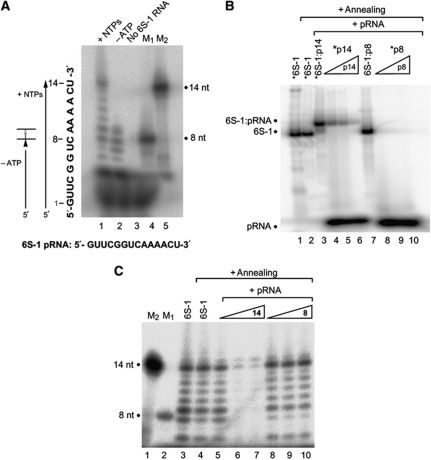
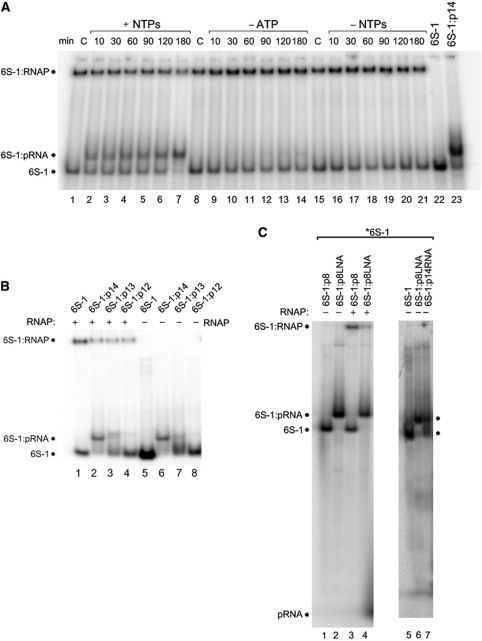
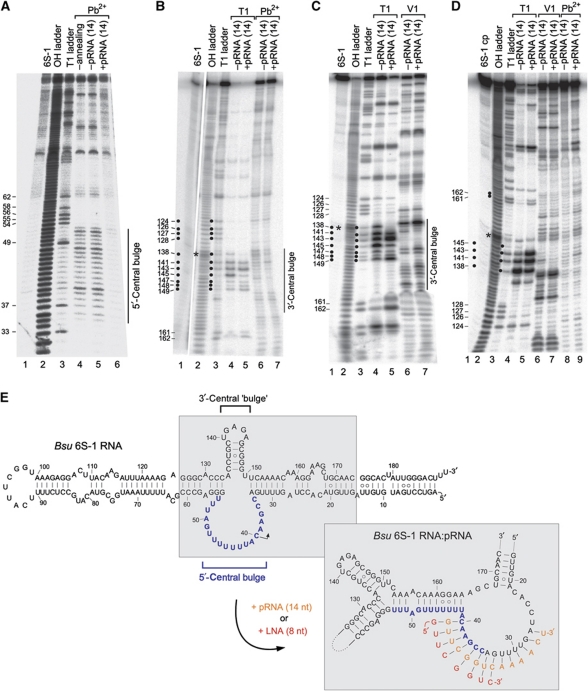
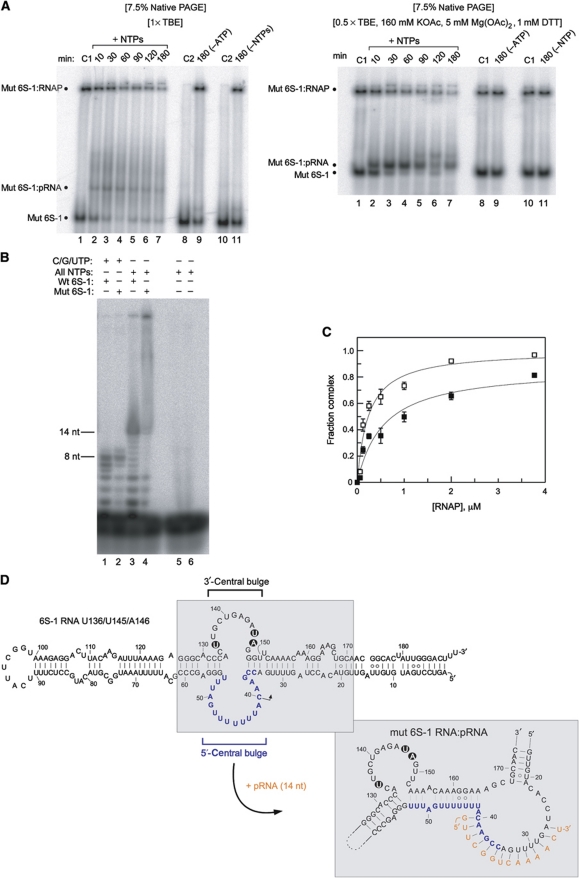
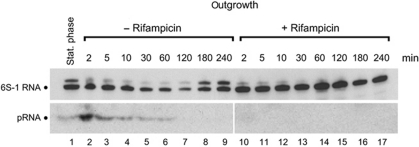
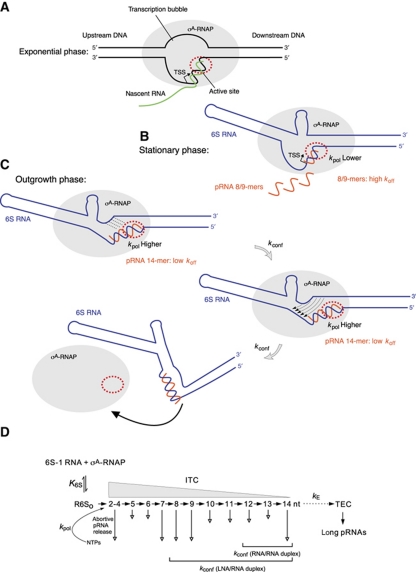
Bacillus subtilis 6S-1 RNA binds to the housekeeping RNA polymerase (σ(A)-RNAP) and directs transcription of short 'product' RNAs (pRNAs). Here, we demonstrate that once newly synthesized pRNAs form a sufficiently stable duplex with 6S-1 RNA, a structural rearrangement is induced in cis, which involves base-pairing between sequences in the 5'-portion of the central bulge and nucleotides that become available as a result of pRNA invasion. The rearrangement decreases 6S-1 RNA affinity for σ(A)-RNAP. Among the pRNA length variants synthesized by σ(A)-RNAP (up to ∼14 nt), only the longer ones, such as 12-14-mers, form a duplex with 6S-1 RNA that is sufficiently long-lived to induce the rearrangement. Yet, an LNA (locked nucleic acid) 8-mer can induce the same rearrangement due to conferring increased duplex stability. We propose that an interplay of rate constants for polymerization (k(pol)), for pRNA:6S-1 RNA hybrid duplex dissociation (k(off)) and for the rearrangement (k(conf)) determines whether pRNAs dissociate or rearrange 6S-1 structure to trigger 6S-1 RNA release from σ(A)-RNAP. A bioinformatic screen suggests that essentially all bacterial 6S RNAs have the potential to undergo a pRNA-induced structural rearrangement.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Ando Y, Asari S, Suzuma S, Yamane K, Nakamura K (2002) Expression of a small RNA, BS203 RNA, from the yocI-yocJ intergenic region of Bacillus subtilis genome. FEMS Microbiol Lett 207: 29–33 - PubMed
-
- Beckmann BM, Burenina OY, Hoch PG, Sharma CM, Kubareva EA, Hartmann RK (2011) In vivo and in vitro analysis of 6S RNA-templated short transcripts in Bacillus subtilis. RNA Biol 8: 839–849 - PubMed
-
- Brownlee GG (1971) Sequence of 6S RNA of E. coli. Nat New Biol 229: 147–149 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
