

Down-regulation of B cell receptor signaling by hematopoietic progenitor kinase 1 (HPK1)-mediated phosphorylation and ubiquitination of activated B cell linker protein (BLNK)
- PMID: 22334673
- PMCID: PMC3322877
- DOI: 10.1074/jbc.M111.310946
Down-regulation of B cell receptor signaling by hematopoietic progenitor kinase 1 (HPK1)-mediated phosphorylation and ubiquitination of activated B cell linker protein (BLNK)
Abstract
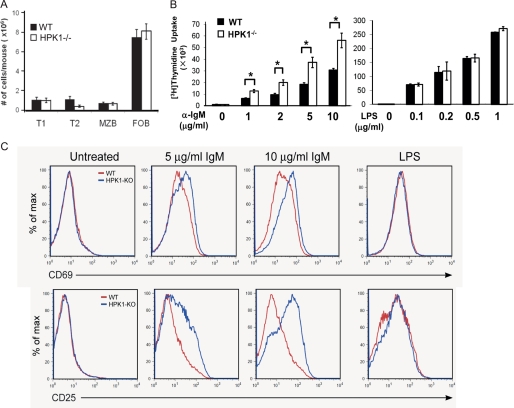
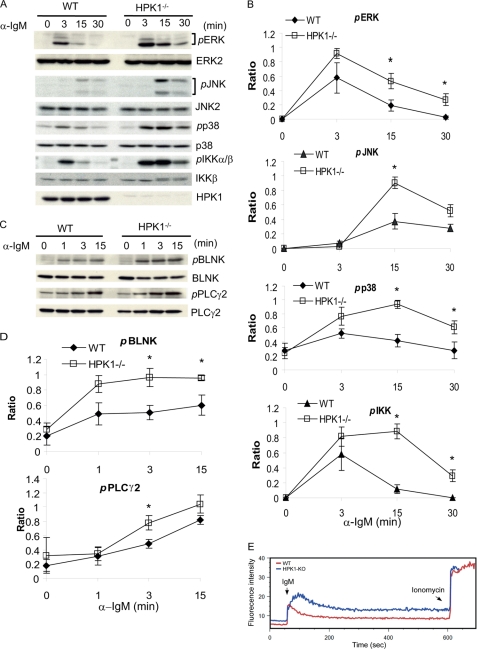
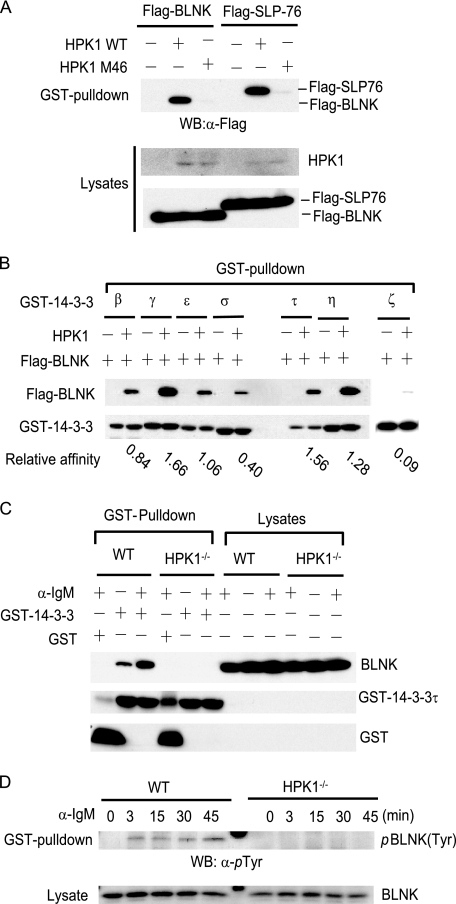
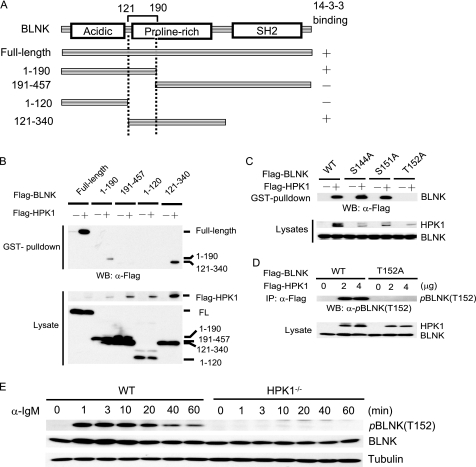
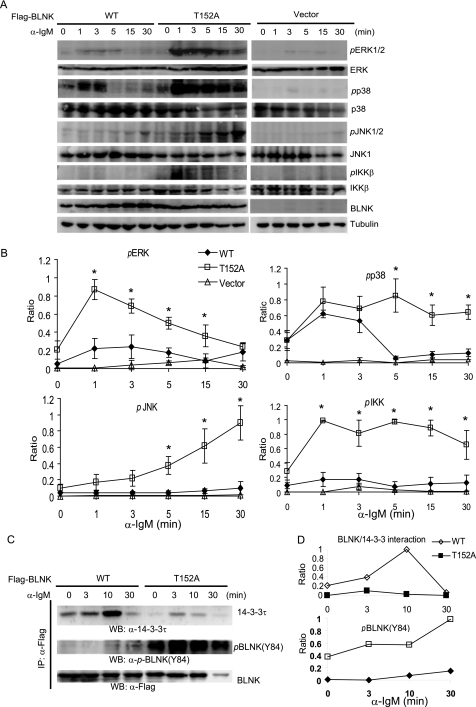
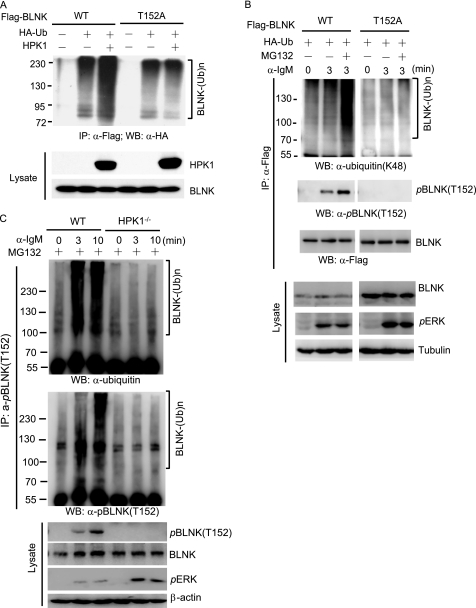
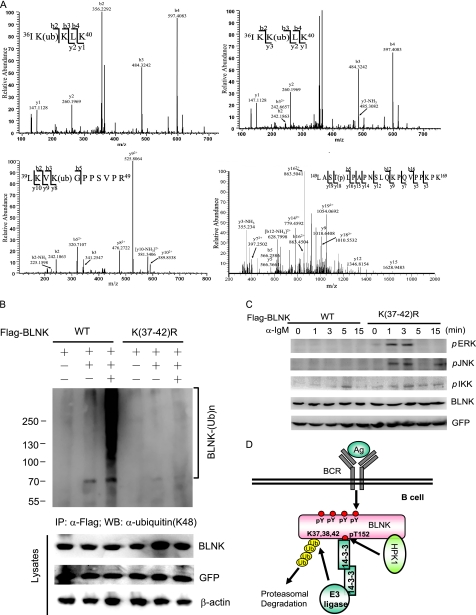
Hematopoietic progenitor kinase 1 (HPK1) is a Ste20-like serine/threonine kinase that suppresses immune responses and autoimmunity. B cell receptor (BCR) signaling activates HPK1 by inducing BLNK/HPK1 interaction. Whether HPK1 can reciprocally regulate BLNK during BCR signaling is unknown. Here, we show that HPK1-deficient B cells display hyper-proliferation and hyper-activation of IκB kinase and MAPKs (ERK, p38, and JNK) upon the ligation of BCR. HPK1 attenuates BCR-induced cell activation via inducing BLNK threonine 152 phosphorylation, which mediates BLNK/14-3-3 binding. Furthermore, threonine 152-phosphorylated BLNK is ubiquitinated at lysine residues 37, 38, and 42, leading to attenuation of MAPK and IκB kinase activation in B cells during BCR signaling. These results reveal a novel negative feedback regulation of BCR signaling by HPK1-mediated phosphorylation, ubiquitination, and subsequent degradation of the activated BLNK.
Figures

References
-
- Yanaba K., Bouaziz J. D., Matsushita T., Magro C. M., St Clair E. W., Tedder T. F. (2008) B-lymphocyte contributions to human autoimmune disease. Immunol. Rev. 223, 284–299 - PubMed
-
- Townsend M. J., Monroe J. G., Chan A. C. (2010) B-cell targeted therapies in human autoimmune diseases: an updated perspective. Immunol. Rev. 237, 264–283 - PubMed
-
- Koretzky G. A., Abtahian F., Silverman M. A. (2006) SLP76 and SLP65. Complex regulation of signaling in lymphocytes and beyond. Nat. Rev. Immunol. 6, 67–78 - PubMed
-
- Datta S. R., Katsov A., Hu L., Petros A., Fesik S. W., Yaffe M. B., Greenberg M. E. (2000) 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol. Cell 6, 41–51 - PubMed
-
- Jang I. K., Zhang J., Gu H. (2009) Grb2, a simple adapter with complex roles in lymphocyte development, function, and signaling. Immunol. Rev. 232, 150–159 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
