

BLANNOTATOR: enhanced homology-based function prediction of bacterial proteins
- PMID: 22335941
- PMCID: PMC3386020
- DOI: 10.1186/1471-2105-13-33
BLANNOTATOR: enhanced homology-based function prediction of bacterial proteins
Abstract
Background: Automated function prediction has played a central role in determining the biological functions of bacterial proteins. Typically, protein function annotation relies on homology, and function is inferred from other proteins with similar sequences. This approach has become popular in bacterial genomics because it is one of the few methods that is practical for large datasets and because it does not require additional functional genomics experiments. However, the existing solutions produce erroneous predictions in many cases, especially when query sequences have low levels of identity with the annotated source protein. This problem has created a pressing need for improvements in homology-based annotation.
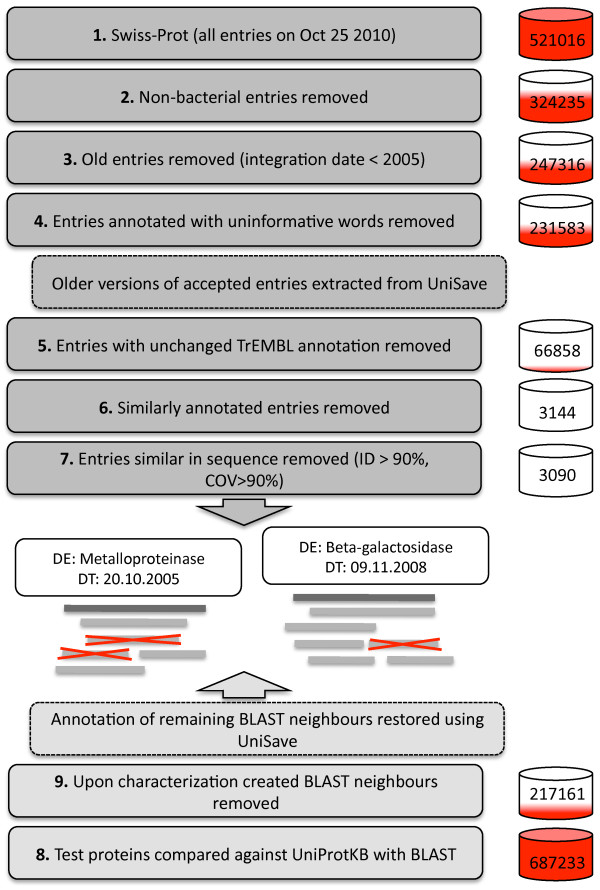
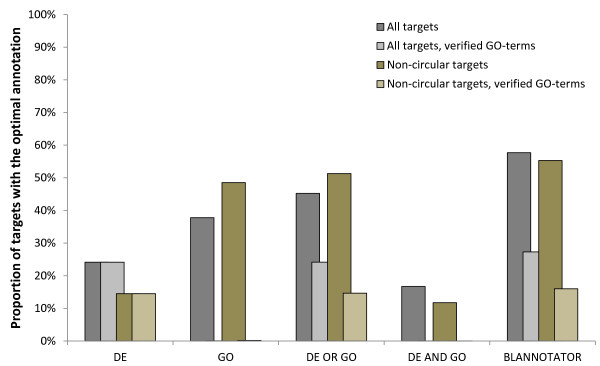
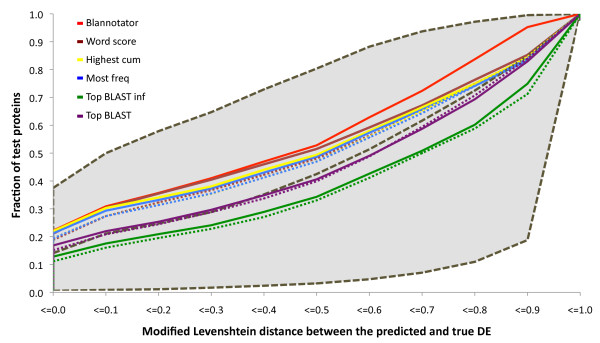
Results: We present an automated method for the functional annotation of bacterial protein sequences. Based on sequence similarity searches, BLANNOTATOR accurately annotates query sequences with one-line summary descriptions of protein function. It groups sequences identified by BLAST into subsets according to their annotation and bases its prediction on a set of sequences with consistent functional information. We show the results of BLANNOTATOR's performance in sets of bacterial proteins with known functions. We simulated the annotation process for 3090 SWISS-PROT proteins using a database in its state preceding the functional characterisation of the query protein. For this dataset, our method outperformed the five others that we tested, and the improved performance was maintained even in the absence of highly related sequence hits. We further demonstrate the value of our tool by analysing the putative proteome of Lactobacillus crispatus strain ST1.
Conclusions: BLANNOTATOR is an accurate method for bacterial protein function prediction. It is practical for genome-scale data and does not require pre-existing sequence clustering; thus, this method suits the needs of bacterial genome and metagenome researchers. The method and a web-server are available at http://ekhidna.biocenter.helsinki.fi/poxo/blannotator/.
Figures



References
-
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Ka-sarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
