

Evaluating pathogenicity of rare variants from dilated cardiomyopathy in the exome era
- PMID: 22337857
- PMCID: PMC3332064
- DOI: 10.1161/CIRCGENETICS.111.961805
Evaluating pathogenicity of rare variants from dilated cardiomyopathy in the exome era
Abstract
Background: Human exome sequencing is a recently developed tool to aid in the discovery of novel coding variants. Now broadly applied, exome sequencing data sets provide a novel opportunity to evaluate the allele frequencies of previously published pathogenic rare variants.
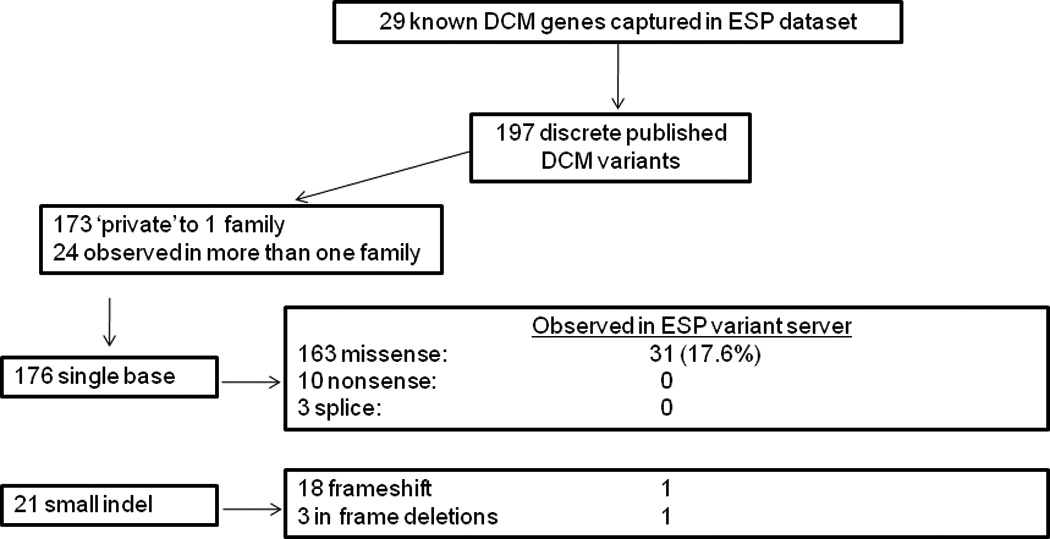
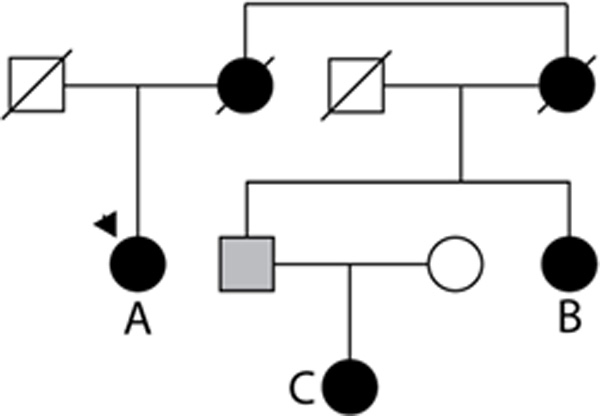
Methods and results: We examined the exome data set from the National Heart, Lung and Blood Institute Exome Sequencing Project and compared this data set with a catalog of 197 previously published rare variants reported as causative of dilated cardiomyopathy (DCM) from familial and sporadic cases. Of these 197, 33 (16.8%) were also present in the Exome Sequencing Project database, raising the question of whether they were uncommon polymorphisms. Supporting functional data has been published for 14 of the 33 (42%), suggesting they are unlikely to be false-positives. The frequencies of these functional variants in the Exome Sequencing Project data set ranged from 0.02 to 1.33% (median 0.04%), which when applied as a cutoff to filter variants in a DCM pedigree identified an additional DCM candidate gene. A greater proportion of sporadic DCM cases had variants that were present in the Exome Sequencing Project data set versus novel variants (ie, not in the Exome Sequencing Project; 44% versus 21%; P=0.002), suggesting some of the variants identified as disease causing in sporadic DCM are either false-positives or low penetrance alleles in human populations.
Conclusions: Rare nonsynonymous variants identified in DCM subjects also present at very low frequencies in public databases are likely relevant for DCM. Allele frequencies >0.04% are of less certain pathogenicity, especially if identified in sporadic cases, although this cutoff should be viewed as preliminary.
Figures
References
-
- Chen JM, Ferec C, Cooper DN. Revealing the human mutome. Clin Genet. 2010;78:310–320. - PubMed
-
- Richards CS, Bale S, Bellissimo DB, Das S, Grody WW, Hegde MR, Lyon E, Ward BE. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med. 2008;10:294–300. - PubMed
-
- Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, Atherton J, Vidaillet HJ, Jr., Spudich S, De Girolami U, Seidman JG, Seidman C, Muntoni F, Muehle G, Johnson W, McDonough B. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–1724. - PubMed
Publication types
MeSH terms
Grants and funding
- RC2 HL102926/HL/NHLBI NIH HHS/United States
- U54 NS065712/NS/NINDS NIH HHS/United States
- 1R01NS072248-01/NS/NINDS NIH HHS/United States
- HL-102924/HL/NHLBI NIH HHS/United States
- HL58626/HL/NHLBI NIH HHS/United States
- HL-102926/HL/NHLBI NIH HHS/United States
- RC2 HL103010/HL/NHLBI NIH HHS/United States
- HL-102923/HL/NHLBI NIH HHS/United States
- RC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102926/HL/NHLBI NIH HHS/United States
- UC2 HL103010/HL/NHLBI NIH HHS/United States
- HL-103010/HL/NHLBI NIH HHS/United States
- RC2 HL102924/HL/NHLBI NIH HHS/United States
- 5U54NS065712-02/NS/NINDS NIH HHS/United States
- UC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102924/HL/NHLBI NIH HHS/United States
- R01 HL058626/HL/NHLBI NIH HHS/United States
- HL-102925/HL/NHLBI NIH HHS/United States
- R01 NS072248/NS/NINDS NIH HHS/United States
- RC2 HL102925/HL/NHLBI NIH HHS/United States
- UC2 HL102925/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
