

Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome
- PMID: 22341451
- PMCID: PMC3343639
- DOI: 10.1016/j.cell.2011.12.035
Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome
Abstract
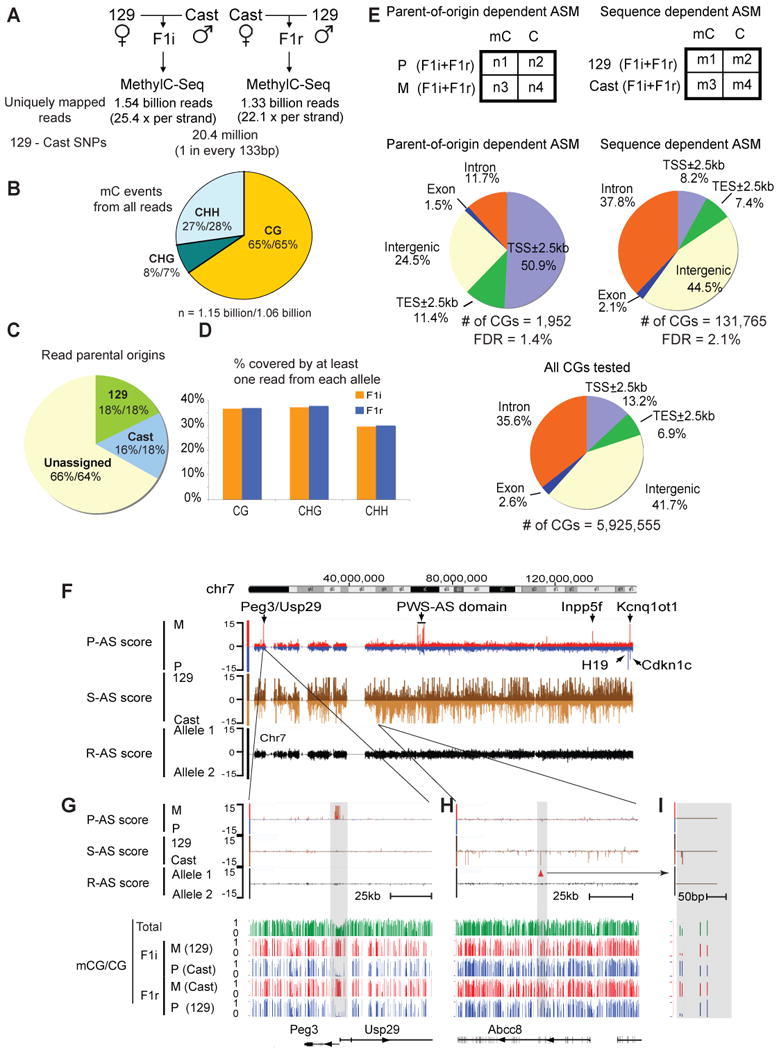
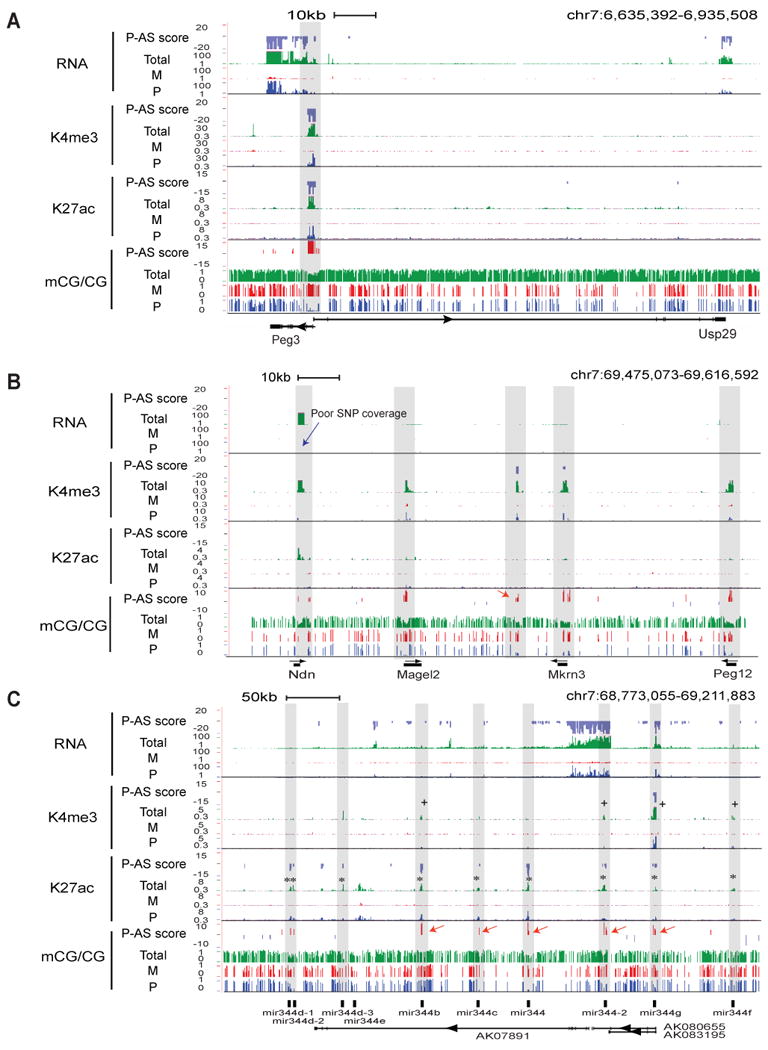
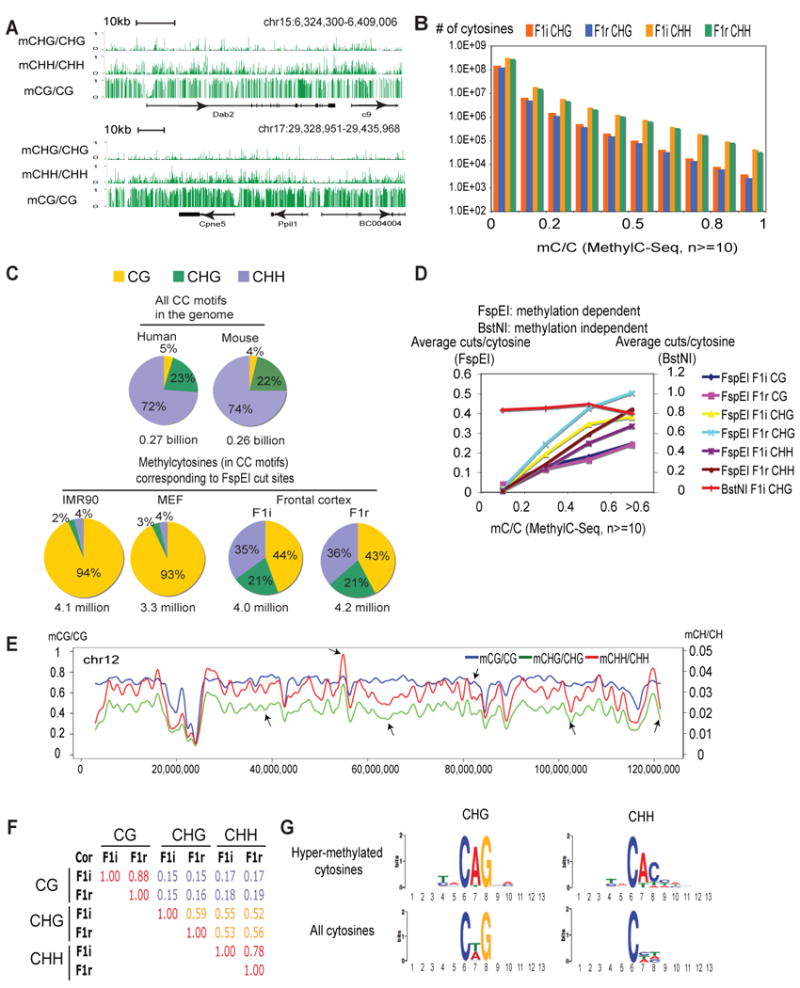
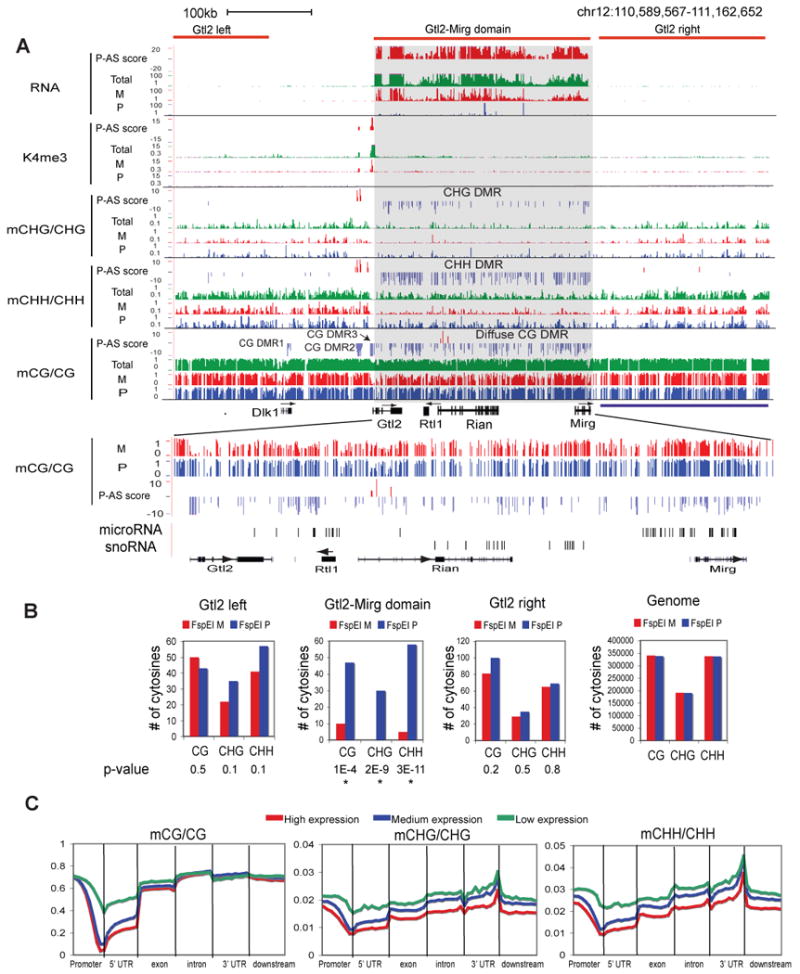
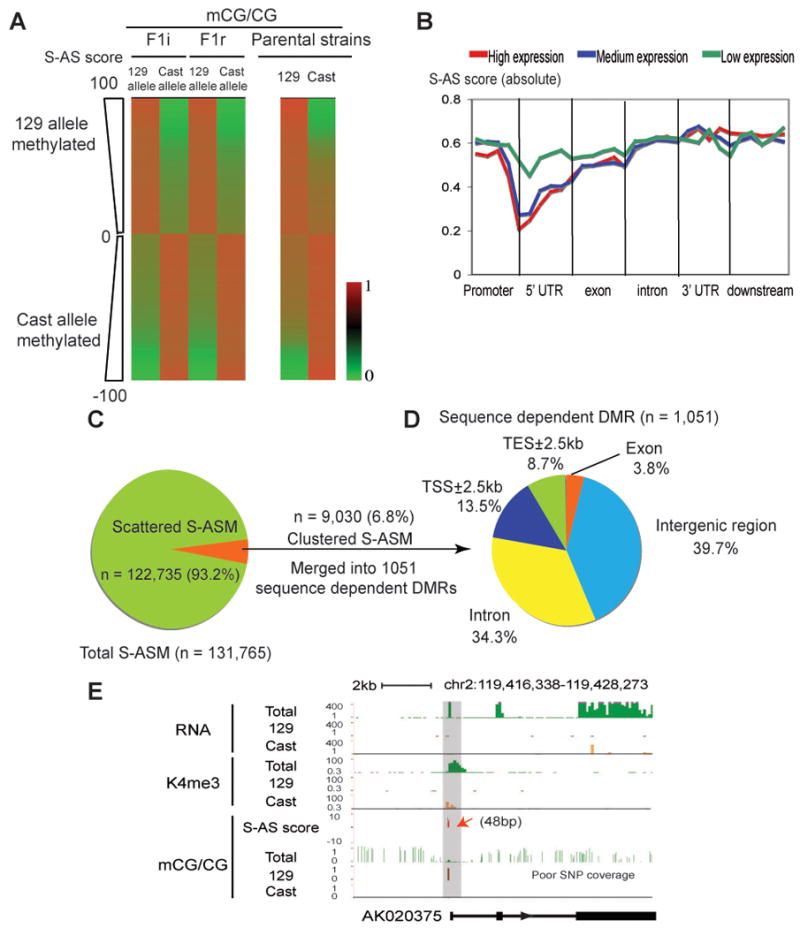
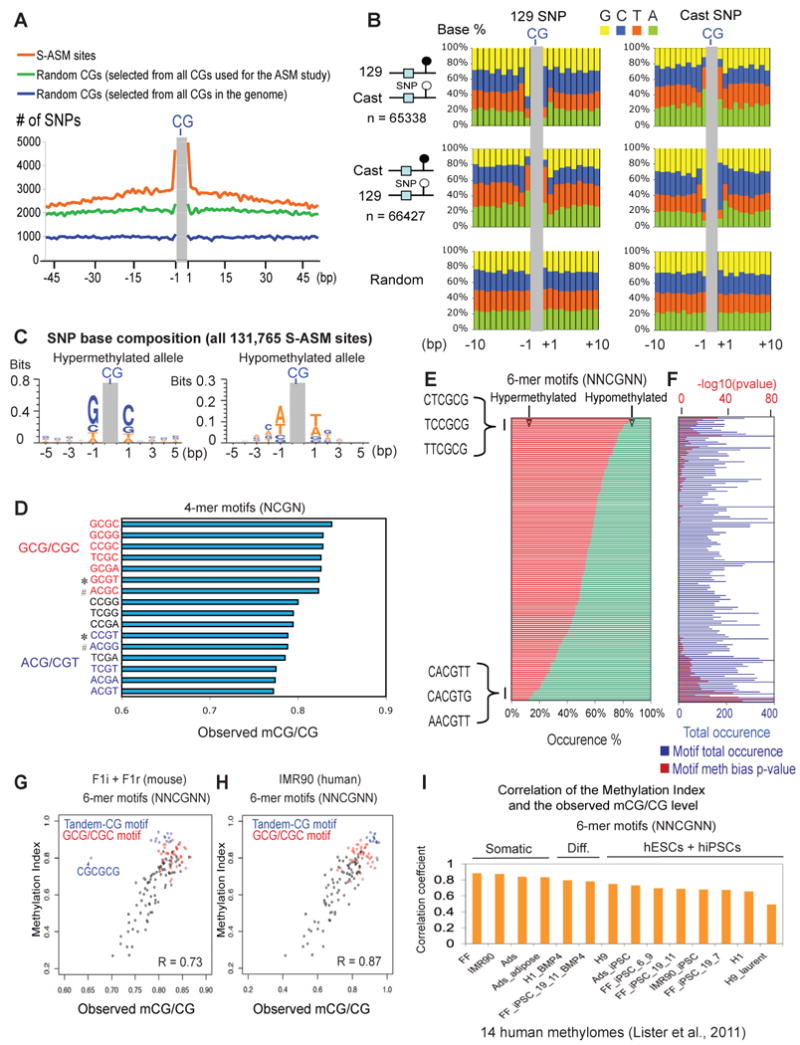
Differential methylation of the two parental genomes in placental mammals is essential for genomic imprinting and embryogenesis. To systematically study this epigenetic process, we have generated a base-resolution, allele-specific DNA methylation (ASM) map in the mouse genome. We find parent-of-origin dependent (imprinted) ASM at 1,952 CG dinucleotides. These imprinted CGs form 55 discrete clusters including virtually all known germline differentially methylated regions (DMRs) and 23 previously unknown DMRs, with some occurring at microRNA genes. We also identify sequence-dependent ASM at 131,765 CGs. Interestingly, methylation at these sites exhibits a strong dependence on the immediate adjacent bases, allowing us to define a conserved sequence preference for the mammalian DNA methylation machinery. Finally, we report a surprising presence of non-CG methylation in the adult mouse brain, with some showing evidence of imprinting. Our results provide a resource for understanding the mechanisms of imprinting and allele-specific gene expression in mammalian cells.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
References
-
- Avila L, Yuen RK, Diego-Alvarez D, Penaherrera MS, Jiang R, Robinson WP. Evaluating DNA methylation and gene expression variability in the human term placenta. Placenta. 2010;31:1070–1077. - PubMed
-
- Babak T, Deveale B, Armour C, Raymond C, Cleary MA, van der Kooy D, Johnson JM, Lim LP. Global survey of genomic imprinting by transcriptome sequencing. Curr Biol. 2008;18:1735–1741. - PubMed
-
- Bird A. DNA methylation patterns and epigenetic memory. Genes & development. 2002;16:6–21. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
