

Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease
- PMID: 22341971
- PMCID: PMC3309204
- DOI: 10.1016/j.ajhg.2012.01.017
Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease
Abstract
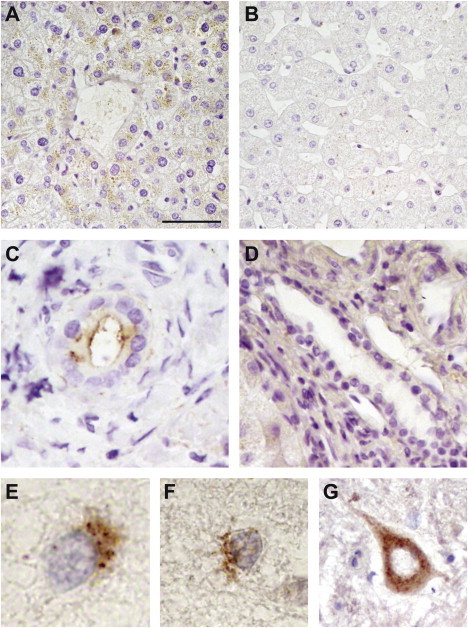
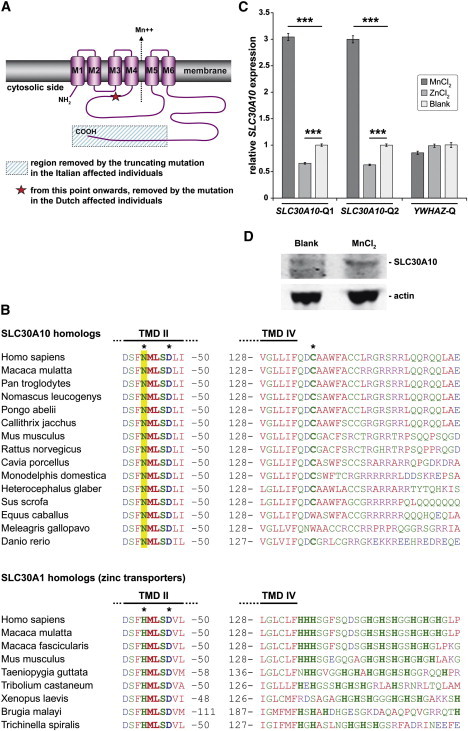
Manganese is essential for several metabolic pathways but becomes toxic in excessive amounts. Manganese levels in the body are therefore tightly regulated, but the responsible protein(s) remain incompletely known. We studied two consanguineous families with neurologic disorders including juvenile-onset dystonia, adult-onset parkinsonism, severe hypermanganesemia, polycythemia, and chronic hepatic disease, including steatosis and cirrhosis. We localized the genetic defect by homozygosity mapping and then identified two different homozygous frameshift SLC30A10 mutations, segregating with disease. SLC30A10 is highly expressed in the liver and brain, including in the basal ganglia. Its encoded protein belongs to a large family of membrane transporters, mediating the efflux of divalent cations from the cytosol. We show the localization of SLC30A10 in normal human liver and nervous system, and its depletion in liver from one affected individual. Our in silico analyses suggest that SLC30A10 possesses substrate specificity different from its closest (zinc-transporting) homologs. We also show that the expression of SLC30A10 and the levels of the encoded protein are markedly induced by manganese in vitro. The phenotype associated with SLC30A10 mutations is broad, including neurologic, hepatic, and hematologic disturbances. Intrafamilial phenotypic variability is also present. Chelation therapy can normalize the manganesemia, leading to marked clinical improvements. In conclusion, we show that SLC30A10 mutations cause a treatable recessive disease with pleomorphic phenotype, and provide compelling evidence that SLC30A10 plays a pivotal role in manganese transport. This work has broad implications for understanding of the manganese biology and pathophysiology in multiple human organs.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures


Comment in
-
A new treatable genetic disorder of manganese metabolism causing dystonia-parkinsonism and cirrhosis: the "new" Wilson's disease?Mov Disord. 2012 Jul;27(8):962. doi: 10.1002/mds.25031. Mov Disord. 2012. PMID: 22807186 No abstract available.
References
-
- Roth J.A. Homeostatic and toxic mechanisms regulating manganese uptake, retention, and elimination. Biol. Res. 2006;39:45–57. - PubMed
-
- Huang C.C., Chu N.S., Lu C.S., Wang J.D., Tsai J.L., Tzeng J.L., Wolters E.C., Calne D.B. Chronic manganese intoxication. Arch. Neurol. 1989;46:1104–1106. - PubMed
-
- Butterworth R.F. Metal toxicity, liver disease and neurodegeneration. Neurotox. Res. 2010;18:100–105. - PubMed
-
- Chalela J.A., Bonillha L., Neyens R., Hays A. Manganese encephalopathy: An under-recognized condition in the intensive care unit. Neurocrit. Care. 2011;14:456–458. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
