

Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons
- PMID: 22342752
- PMCID: PMC3313683
- DOI: 10.1016/j.cub.2012.02.005
Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons
Abstract
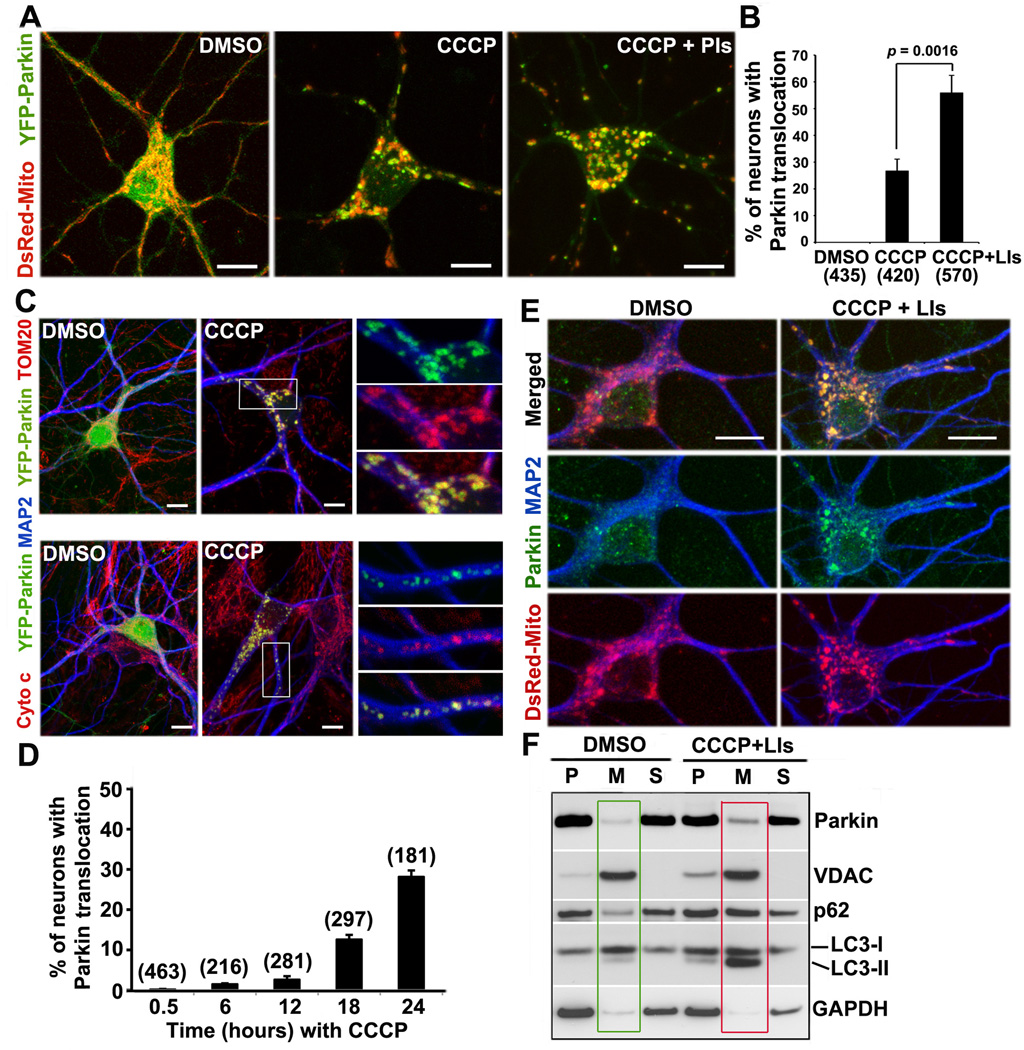
Mitochondria are essential for neuronal survival and function. Proper degradation of aged and damaged mitochondria through mitophagy is a key cellular pathway for mitochondrial quality control. Recent studies have indicated that PINK1/Parkin-mediated pathways ensure mitochondrial integrity and function. Translocation of Parkin to damaged mitochondria induces mitophagy in many nonneuronal cell types. However, evidence showing Parkin translocation in primary neurons is controversial, leaving unanswered questions as to how and where Parkin-mediated mitophagy occurs in neurons. Here, we report the unique process of dissipating mitochondrial Δψ(m)-induced and Parkin-mediated mitophagy in mature cortical neurons. Compared with nonneuronal cells, neuronal mitophagy is a much slower and compartmentally restricted process, coupled with reduced anterograde mitochondrial transport. Parkin-targeted mitochondria are accumulated in the somatodendritic regions where mature lysosomes are predominantly located. Time-lapse imaging shows dynamic formation and elimination of Parkin- and LC3-ring-like structures surrounding depolarized mitochondria through the autophagy-lysosomal pathway in the soma. Knocking down Parkin in neurons impairs the elimination of dysfunctional mitochondria. Thus, our study provides neuronal evidence for dynamic and spatial Parkin-mediated mitophagy, which will help us understand whether altered mitophagy contributes to pathogenesis of several major neurodegenerative diseases characterized by mitochondrial dysfunction and impaired transport.
Copyright © 2012 Elsevier Ltd. All rights reserved.
Figures



References
-
- Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 2004;279:18614–18622. - PubMed
-
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
