

Identification of microRNA-regulated gene networks by expression analysis of target genes
- PMID: 22345618
- PMCID: PMC3371699
- DOI: 10.1101/gr.130435.111
Identification of microRNA-regulated gene networks by expression analysis of target genes
Abstract
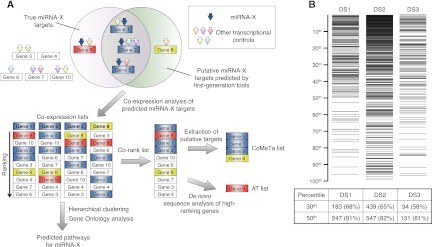
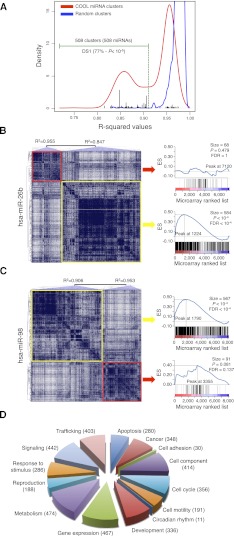
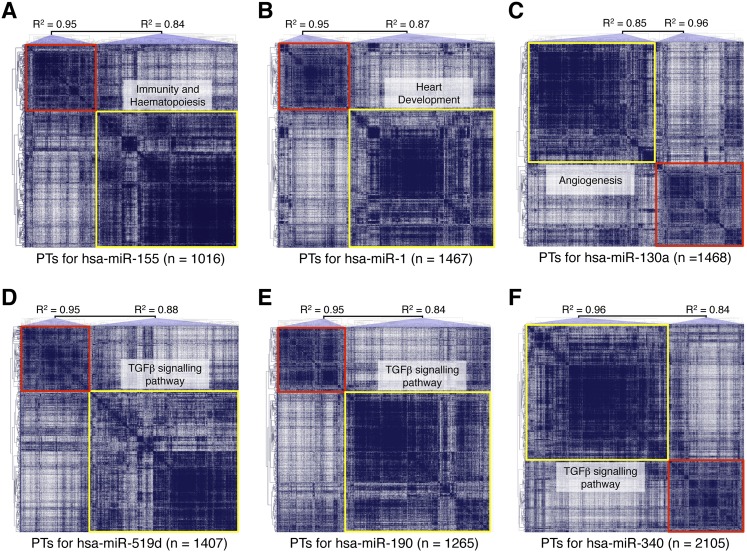
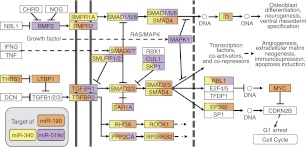
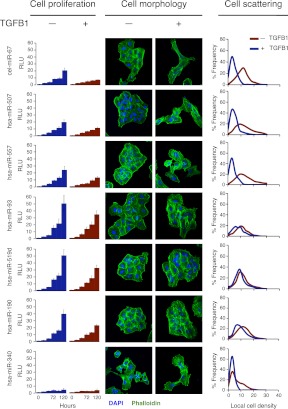
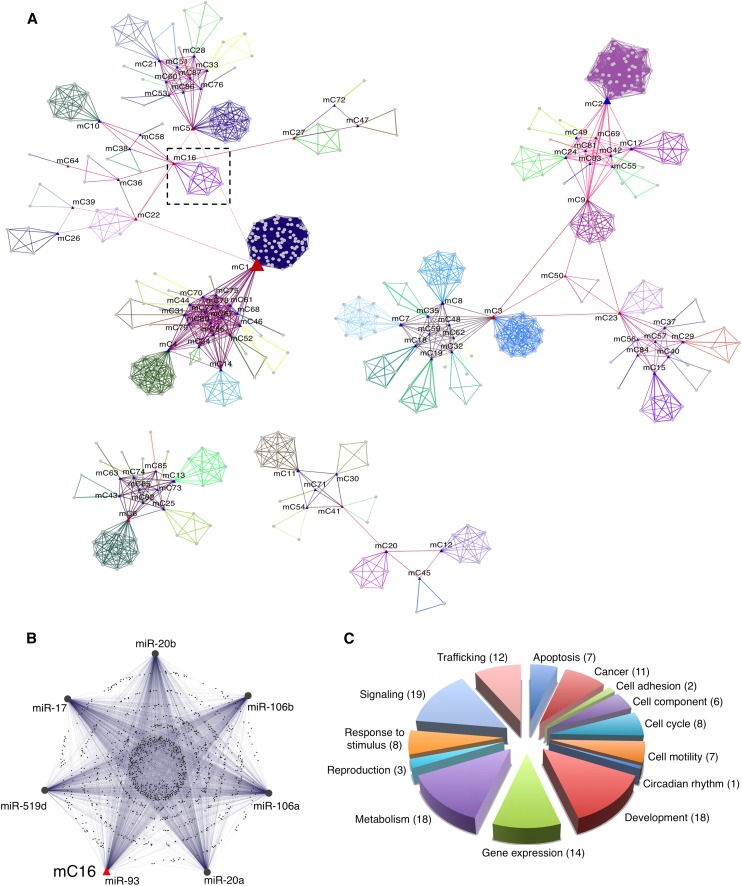
MicroRNAs (miRNAs) and transcription factors control eukaryotic cell proliferation, differentiation, and metabolism through their specific gene regulatory networks. However, differently from transcription factors, our understanding of the processes regulated by miRNAs is currently limited. Here, we introduce gene network analysis as a new means for gaining insight into miRNA biology. A systematic analysis of all human miRNAs based on Co-expression Meta-analysis of miRNA Targets (CoMeTa) assigns high-resolution biological functions to miRNAs and provides a comprehensive, genome-scale analysis of human miRNA regulatory networks. Moreover, gene cotargeting analyses show that miRNAs synergistically regulate cohorts of genes that participate in similar processes. We experimentally validate the CoMeTa procedure through focusing on three poorly characterized miRNAs, miR-519d/190/340, which CoMeTa predicts to be associated with the TGFβ pathway. Using lung adenocarcinoma A549 cells as a model system, we show that miR-519d and miR-190 inhibit, while miR-340 enhances TGFβ signaling and its effects on cell proliferation, morphology, and scattering. Based on these findings, we formalize and propose co-expression analysis as a general paradigm for second-generation procedures to recognize bona fide targets and infer biological roles and network communities of miRNAs.
Figures
References
-
- Betel D, Wilson M, Gabow A, Marks DS, Sander C 2008. The microRNA.org resource: Targets and expression. Nucleic Acids Res 36: D149–D153 - PMC - PubMed
-
- Bushati N, Cohen SM 2007. microRNA functions. Annu Rev Cell Dev Biol 23: 175–205 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources