

In situ assay of fatty acid β-oxidation by metabolite profiling following permeabilization of cell membranes
- PMID: 22345709
- PMCID: PMC3329378
- DOI: 10.1194/jlr.D022608
In situ assay of fatty acid β-oxidation by metabolite profiling following permeabilization of cell membranes
Abstract
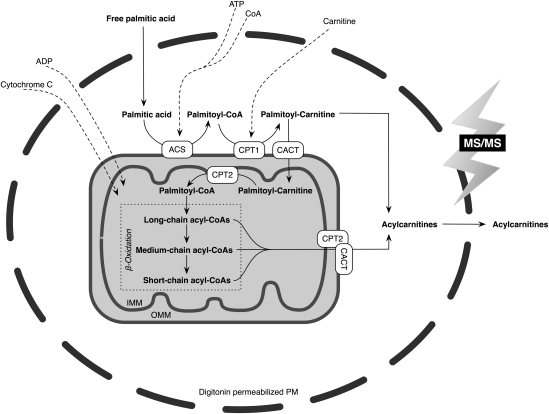
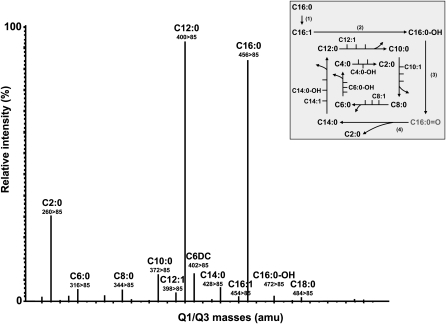
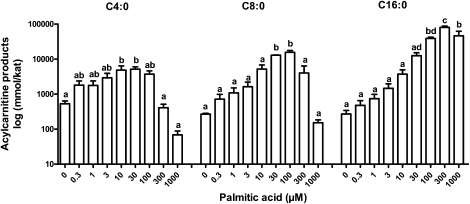
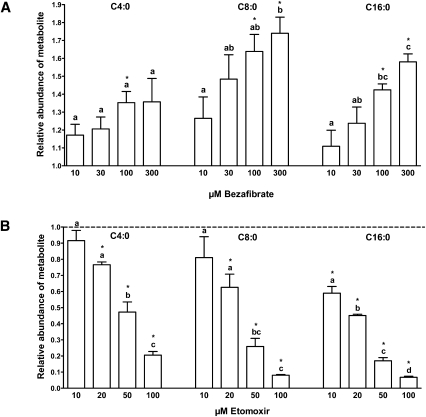
Quantitative analysis of mitochondrial FA β-oxidation (FAO) has drawn increasing interest for defining lipid-induced metabolic dysfunctions, such as in obesity-induced insulin resistance, and evaluating pharmacologic strategies to improve β-oxidation function. The aim was to develop a new assay to quantify β-oxidation function in intact mitochondria and with a low amount of cell material. Cell membranes of primary human fibroblasts were permeabilized with digitonin prior to a load with FFA substrate. Following 120 min of incubation, the various generated acylcarnitines were extracted from both cells and incubation medium by protein precipitation/desalting and subjected to solid-phase extraction. A panel of 30 acylcarnitines per well was quantified by MS/MS and normalized to citrate synthase activity to analyze mitochondrial metabolite flux. Pretreatment with bezafibrate and etomoxir revealed stimulating and inhibiting regulatory effects on β-oxidation function, respectively. In addition to the advantage of a much shorter assay time due to in situ permeabilization compared with whole-cell incubation systems, the method allows the detection of multiple acylcarnitines from an only limited amount of intact cells, particularly relevant to the use of primary cells. This novel approach facilitates highly sensitive, simple, and fast monitoring of pharmacological effects on FAO.
Figures
References
-
- Matern D., Cuthbert C. D., Tortorelli S., Rinaldo P.2008. Inherited abnormalities in mitochondrial fatty acid oxidation. In Walker's Pediatric Gastrointestinal Disease. R. E. Kleinman, O. J. Goulet, G. M. Vergani, et al., editors. BC Decker, Inc., Hamilton, Ontario. 1287–1307.
-
- Muoio D. M., Newgard C. B. 2008. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat. Rev. Mol. Cell Biol. 9: 193–205 - PubMed
-
- Koves T. R., Ussher J. R., Noland R. C., Slentz D., Mosedale M., Ilkayeva O., Bain J., Stevens R., Dyck J. R., Newgard C. B., et al. 2008. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 7: 45–56 - PubMed
-
- Kølvraa S., Gregersen N., Christensen E., Hobolth N. 1982. In vitro fibroblast studies in a patient with C6-C10-dicarboxylic aciduria: evidence for a defect in general acyl-CoA dehydrogenase. Clin. Chim. Acta. 126: 53–67 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
