

Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders
- PMID: 22346768
- PMCID: PMC3276563
- DOI: 10.1371/journal.pgen.1002521
Genetic and functional analyses of SHANK2 mutations suggest a multiple hit model of autism spectrum disorders
Abstract
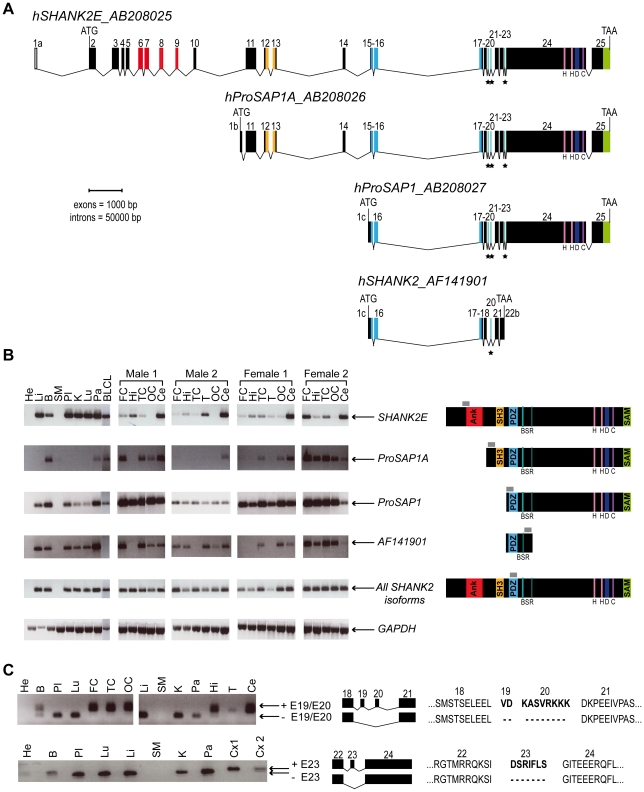
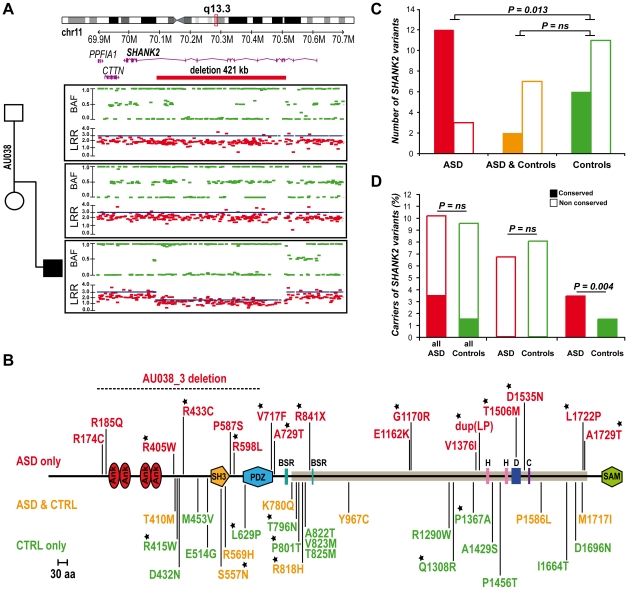
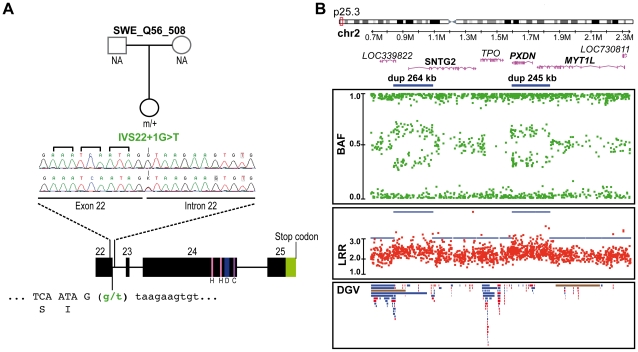
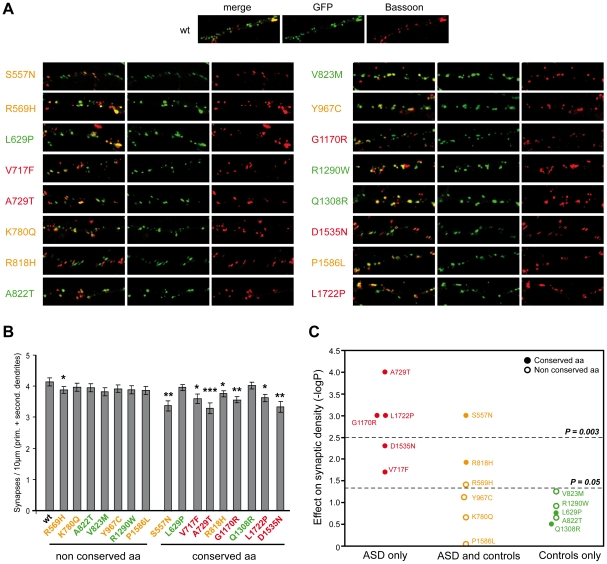
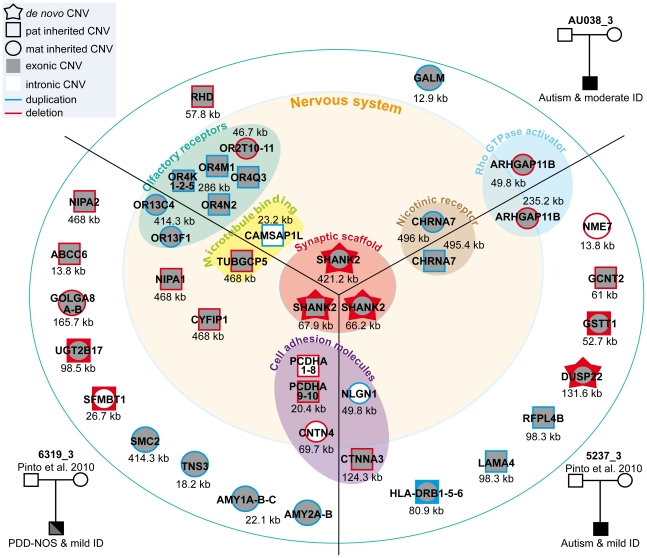
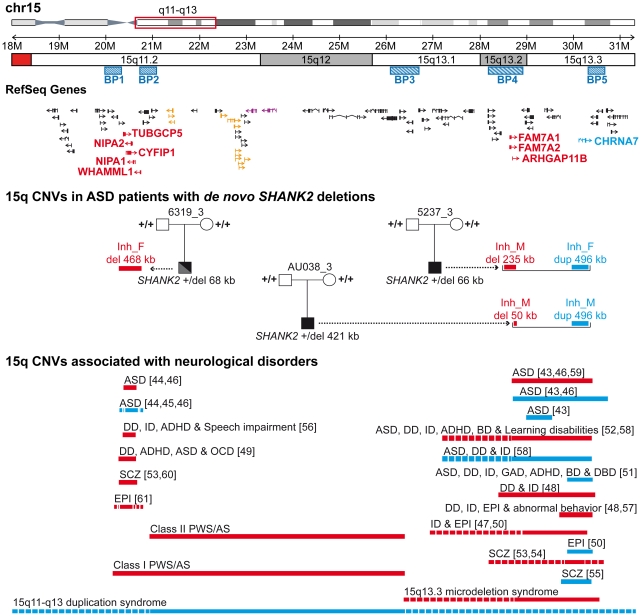
Autism spectrum disorders (ASD) are a heterogeneous group of neurodevelopmental disorders with a complex inheritance pattern. While many rare variants in synaptic proteins have been identified in patients with ASD, little is known about their effects at the synapse and their interactions with other genetic variations. Here, following the discovery of two de novo SHANK2 deletions by the Autism Genome Project, we identified a novel 421 kb de novo SHANK2 deletion in a patient with autism. We then sequenced SHANK2 in 455 patients with ASD and 431 controls and integrated these results with those reported by Berkel et al. 2010 (n = 396 patients and n = 659 controls). We observed a significant enrichment of variants affecting conserved amino acids in 29 of 851 (3.4%) patients and in 16 of 1,090 (1.5%) controls (P = 0.004, OR = 2.37, 95% CI = 1.23-4.70). In neuronal cell cultures, the variants identified in patients were associated with a reduced synaptic density at dendrites compared to the variants only detected in controls (P = 0.0013). Interestingly, the three patients with de novo SHANK2 deletions also carried inherited CNVs at 15q11-q13 previously associated with neuropsychiatric disorders. In two cases, the nicotinic receptor CHRNA7 was duplicated and in one case the synaptic translation repressor CYFIP1 was deleted. These results strengthen the role of synaptic gene dysfunction in ASD but also highlight the presence of putative modifier genes, which is in keeping with the "multiple hit model" for ASD. A better knowledge of these genetic interactions will be necessary to understand the complex inheritance pattern of ASD.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 4th Ed. American Psychiatric Press, Washington D.C; 1994.
-
- Fernell E, Gillberg C. Autism spectrum disorder diagnoses in Stockholm preschoolers. Res Dev Disabil. 2010;31:680–685. - PubMed
-
- Freitag CM. The genetics of autistic disorders and its clinical relevance: a review of the literature. Mol Psychiatry. 2007;12:2–22. - PubMed
-
- Bourgeron T. A synaptic trek to autism. Curr Opin Neurobiol. 2009;19:231–234. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
