

Virus identification in unknown tropical febrile illness cases using deep sequencing
- PMID: 22347512
- PMCID: PMC3274504
- DOI: 10.1371/journal.pntd.0001485
Virus identification in unknown tropical febrile illness cases using deep sequencing
Abstract
Dengue virus is an emerging infectious agent that infects an estimated 50-100 million people annually worldwide, yet current diagnostic practices cannot detect an etiologic pathogen in ∼40% of dengue-like illnesses. Metagenomic approaches to pathogen detection, such as viral microarrays and deep sequencing, are promising tools to address emerging and non-diagnosable disease challenges. In this study, we used the Virochip microarray and deep sequencing to characterize the spectrum of viruses present in human sera from 123 Nicaraguan patients presenting with dengue-like symptoms but testing negative for dengue virus. We utilized a barcoding strategy to simultaneously deep sequence multiple serum specimens, generating on average over 1 million reads per sample. We then implemented a stepwise bioinformatic filtering pipeline to remove the majority of human and low-quality sequences to improve the speed and accuracy of subsequent unbiased database searches. By deep sequencing, we were able to detect virus sequence in 37% (45/123) of previously negative cases. These included 13 cases with Human Herpesvirus 6 sequences. Other samples contained sequences with similarity to sequences from viruses in the Herpesviridae, Flaviviridae, Circoviridae, Anelloviridae, Asfarviridae, and Parvoviridae families. In some cases, the putative viral sequences were virtually identical to known viruses, and in others they diverged, suggesting that they may derive from novel viruses. These results demonstrate the utility of unbiased metagenomic approaches in the detection of known and divergent viruses in the study of tropical febrile illness.
Conflict of interest statement
The authors have declared that no competing interests exist.
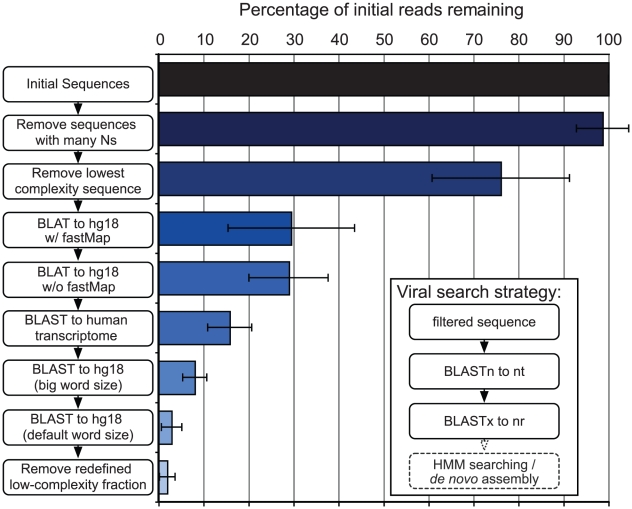
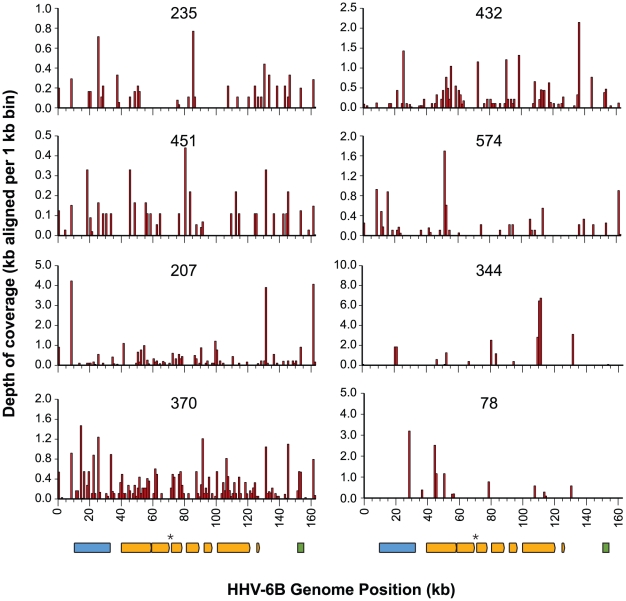
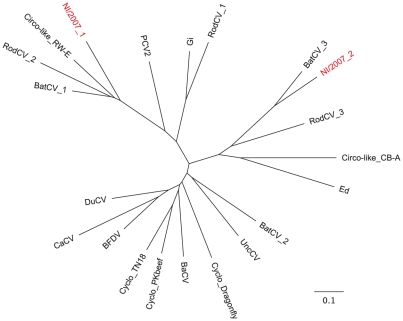
Figures
References
-
- Mabey D, Peeling RW, Ustianowski A, Perkins MD. Tropical infectious diseases: Diagnostics for the developing world. Nat Rev Micro. 2004;2:231–240. doi: 10.1038/nrmicro841. - DOI - PubMed
-
- Dong J, Olano JP, McBride JW, Walker DH. Emerging Pathogens: Challenges and Successes of Molecular Diagnostics. J Mol Diagn. 2008;10:185–197. doi: 10.2353/jmoldx.2008.070063. - DOI - PMC - PubMed
-
- Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, et al. Global trends in emerging infectious diseases. Nature. 2008;451:990–993. doi: 10.1038/nature06536. - DOI - PMC - PubMed
-
- World Health Organization. Dengue haemorrhagic fever: diagnosis, treatment, prevention and control. 2nd edition. Geneva: World Health Organization; 1997. Available: http://www.who.int/csr/resources/publications/dengue/Denguepublication/en/. Accessed 27 Feb 2011.
-
- Kyle JL, Harris E. Global spread and persistence of dengue. Annu Rev Microbiol. 2008;62:71–92. doi: 10.1146/annurev.micro.62.081307.163005. - DOI - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
