

Inflammation in atherosclerosis: a cause or a result of vascular disorders?
- PMID: 22348535
- PMCID: PMC3822968
- DOI: 10.1111/j.1582-4934.2012.01552.x
Inflammation in atherosclerosis: a cause or a result of vascular disorders?
Abstract
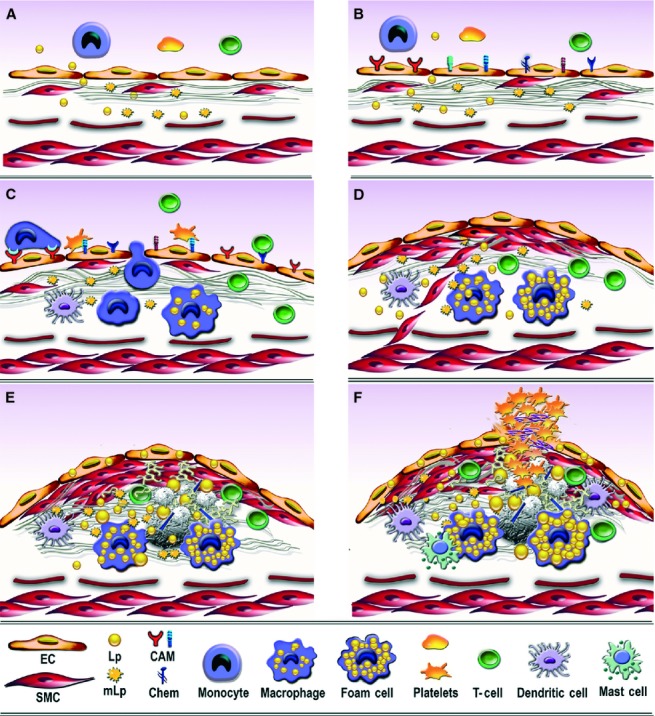
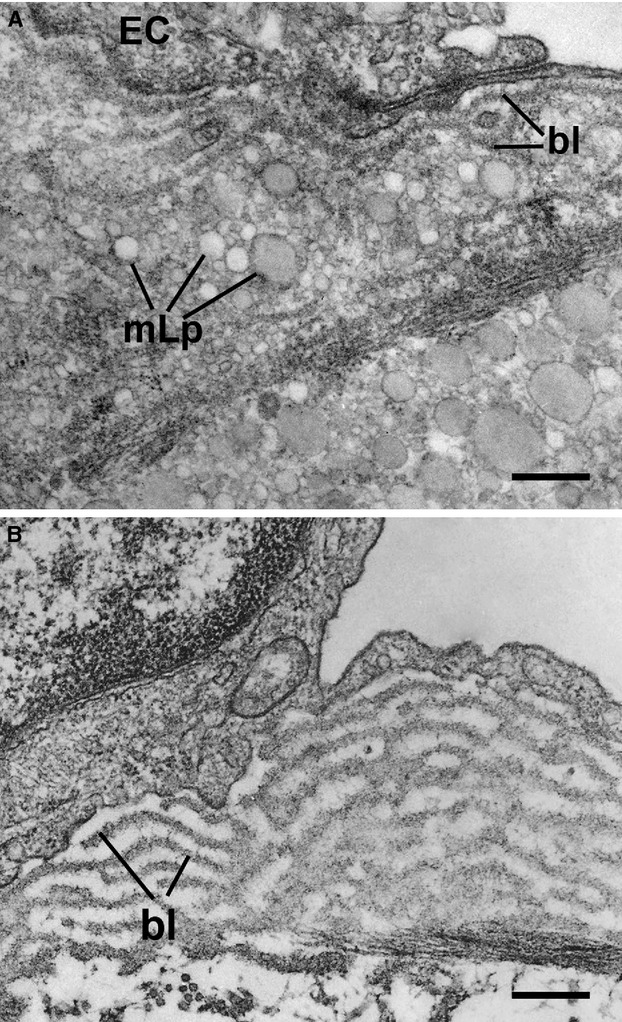
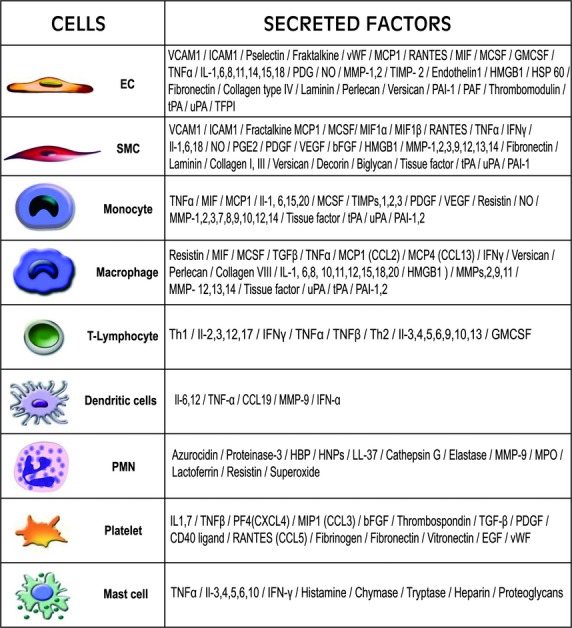
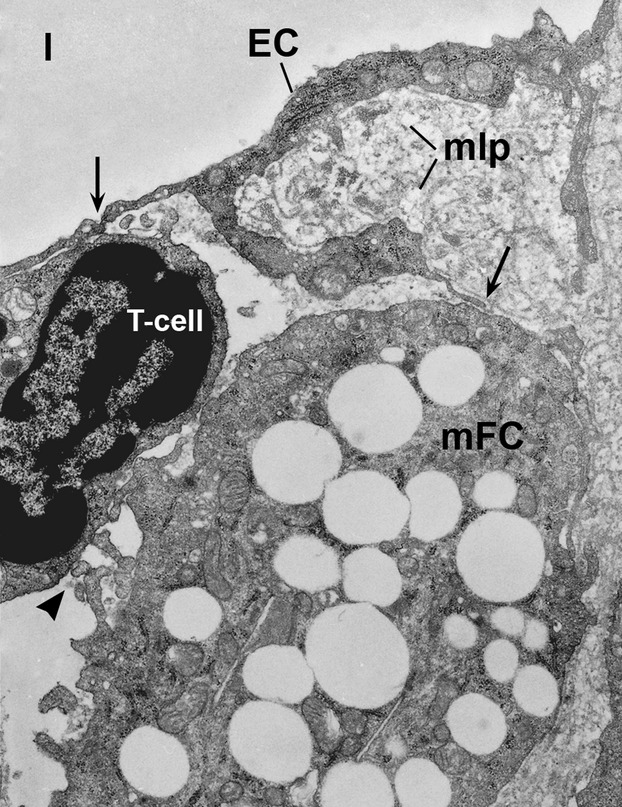
Sound data support the concept that in atherosclerosis, inflammation and dyslipidemia intersect each other and that irrespective of the initiator, both participate from the early stages to the ultimate fate of the atheromatous plaque. The two partakers manoeuvre a vicious circle in atheroma formation: dyslipidaemia triggers an inflammatory process and inflammation elicits dyslipidaemia. Independent of the initial cause, the atherosclerotic lesions occur focally, in particular arterial-susceptible sites, by a process that, although continuous, can be arbitrarily divided into a sequence of consecutive stages that lead from fatty streak to the fibro-lipid plaque and ultimately to plaque rupture and thrombosis. In the process, the initial event is a change in endothelial cells (EC) constitutive properties. Then, the molecular alarm signals send by dysfunctional EC are decoded by specific blood immune cells (monocytes, T lymphocytes, neutrophils, mast cells) and by the resident vascular cells, that respond by initiating a robust inflammatory process, in which the cells and the factors they secrete hasten the atheroma development. Direct and indirect crosstalk between the cells housed within the nascent plaque, complemented by the increase in risk factors of atherosclerosis lead to atheroma development and outcome. The initial inflammatory response can be regarded as a defense/protective reaction mechanism, but its further amplification, speeds up atherosclerosis. In this review, we provide an overview on the role of inflammation and dyslipidaemia and their intersection in atherogenesis. The data may add to the foundation of a novel attitude in the diagnosis and treatment of atherosclerosis.
© 2012 The Authors Journal of Cellular and Molecular Medicine © 2012 Foundation for Cellular and Molecular Medicine/Blackwell Publishing Ltd.
Figures
References
-
- Millonig G, Malcom GT, Wick G. Early inflammatory-immunological lesions in juvenile atherosclerosis from the pathobiological determinants of atherosclerosis in youth (PDAY)-study. Atherosclerosis. 2002;160:441–8. - PubMed
-
- Tsimikas S, Willerson JT, Ridker PM. C-reactive protein and other emerging blood biomarkers to optimize risk stratification of vulnerable patients. J Am Coll Cardiol. 2006;47:C19–31. - PubMed
-
- VanderLaan PA, Reardon CA, Getz GS. Site-specificity of atherosclerosis: site-selective responses to atherosclerotic modulators. Arterioscler Thromb Vasc Biol. 2004;24:12–22. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
