

Adenosine 5-diphosphate-ribose is a neural regulator in primate and murine large intestine along with β-NAD(+)
- PMID: 22351627
- PMCID: PMC3573313
- DOI: 10.1113/jphysiol.2011.222414
Adenosine 5-diphosphate-ribose is a neural regulator in primate and murine large intestine along with β-NAD(+)
Abstract
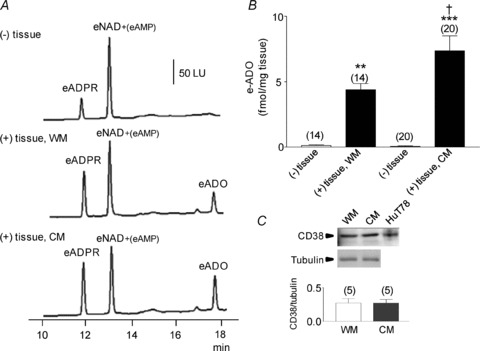
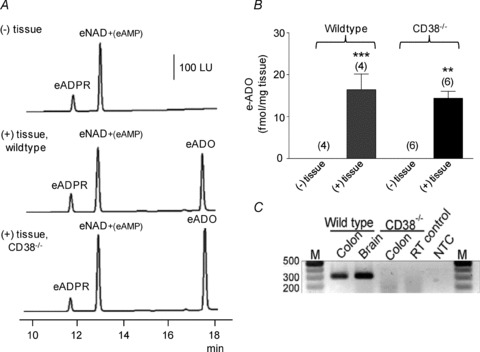
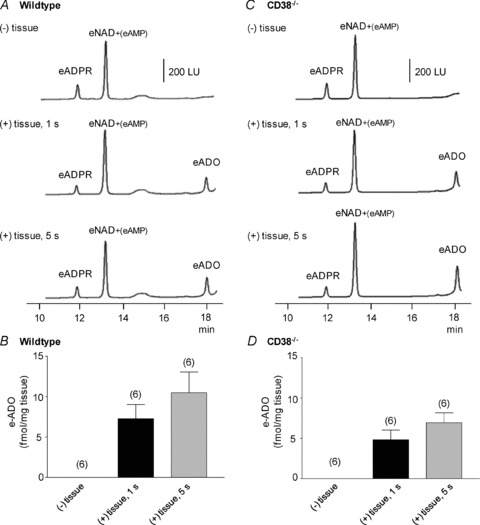
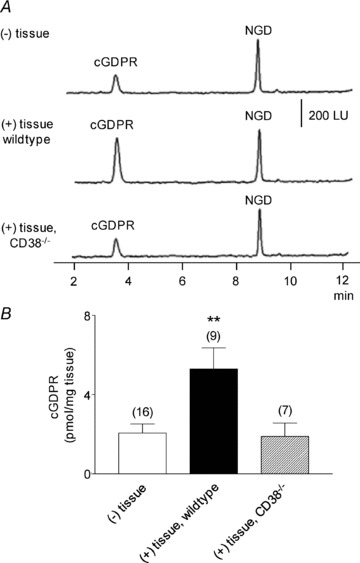
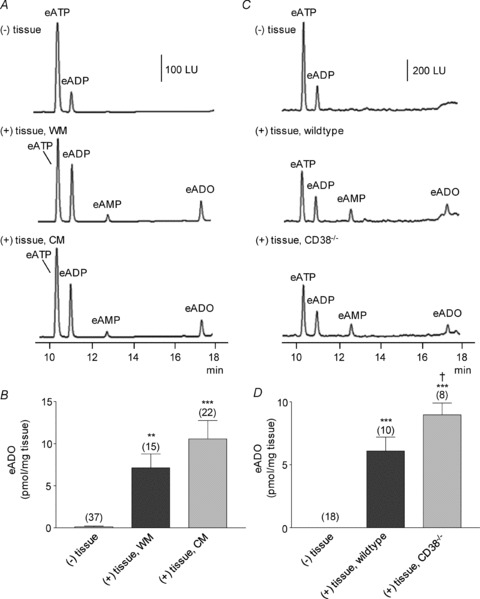
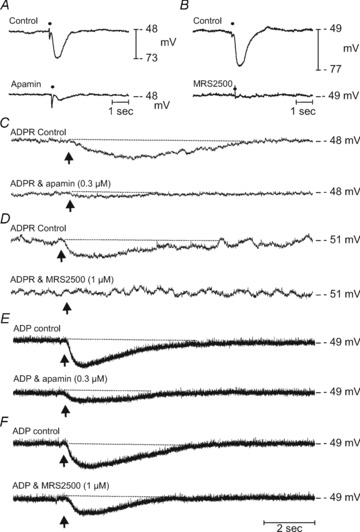
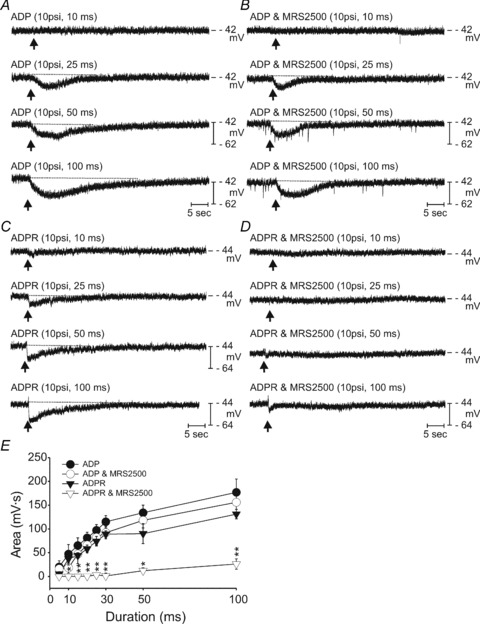
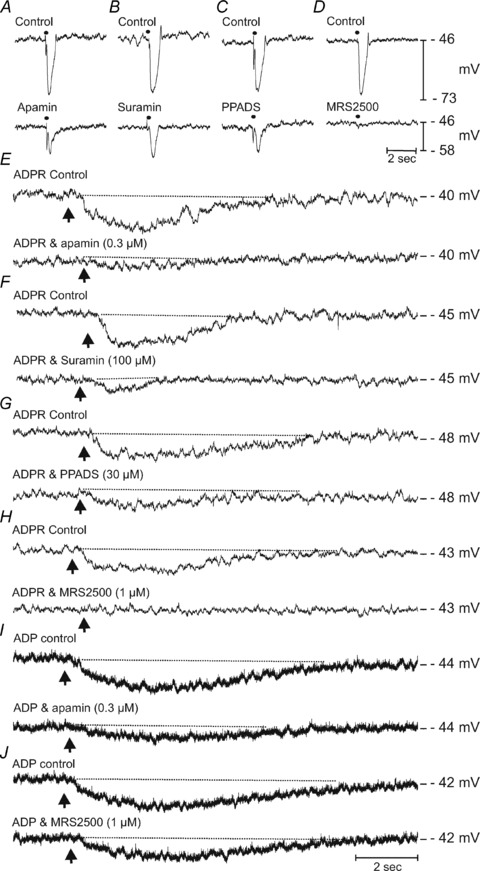
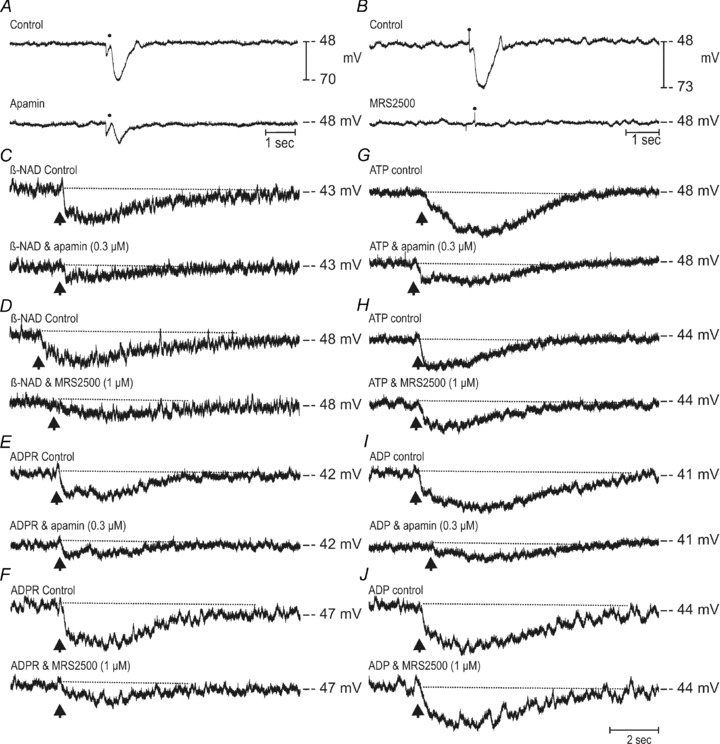
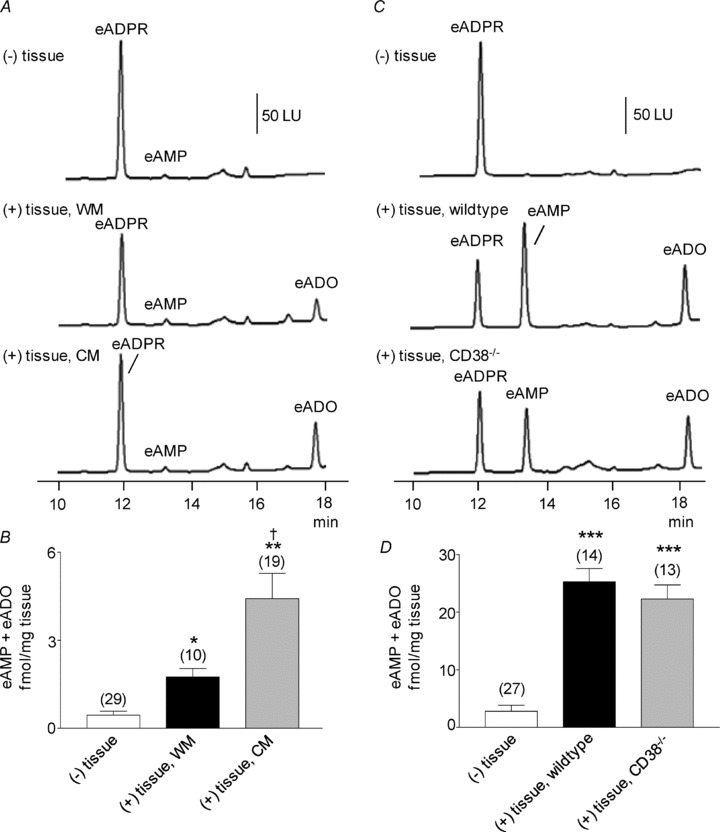
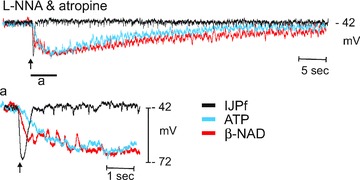
Adenosine 5′-triphosphate (ATP) has long been considered to be the purine inhibitory neurotransmitter in gastrointestinal (GI) muscles, but recent studies indicate that another purine nucleotide, β-nicotinamide adenine dinucleotide (β-NAD(+)), meets pre- and postsynaptic criteria for a neurotransmitter better than ATP in primate and murine colons. Using a small-volume superfusion assay and HPLC with fluorescence detection and intracellular microelectrode techniques we compared β-NAD(+) and ATP metabolism and postjunctional effects of the primary extracellular metabolites of β-NAD(+) and ATP, namely ADP-ribose (ADPR) and ADP in colonic muscles from cynomolgus monkeys and wild-type (CD38(+/+)) and CD38(−/−) mice. ADPR and ADP caused membrane hyperpolarization that, like nerve-evoked inhibitory junctional potentials (IJPs), were inhibited by apamin. IJPs and hyperpolarization responses to ADPR, but not ADP, were inhibited by the P2Y1 receptor antagonist (1R,2S,4S,5S)-4-[2-iodo-6-(methylamino)-9H-purin-9-yl]-2-(phosphonooxy)bicyclo[3.1.0]hexane-1-methanol dihydrogen phosphate ester tetraammonium salt (MRS2500). Degradation of β-NAD(+) and ADPR was greater per unit mass in muscles containing only nerve processes than in muscles also containing myenteric ganglia. Thus, mechanisms for generation of ADPR from β-NAD(+) and for termination of the action of ADPR are likely to be present near sites of neurotransmitter release. Degradation of β-NAD(+) to ADPR and other metabolites appears to be mediated by pathways besides CD38, the main NAD-glycohydrolase in mammals. Degradation of β-NAD(+) and ATP were equal in colon. ADPR like its precursor, β-NAD(+), mimicked the effects of the endogenous purine neurotransmitter in primate and murine colons. Taken together, our observations support a novel hypothesis in which multiple purines contribute to enteric inhibitory regulation of gastrointestinal motility.
Figures
Similar articles
-
β-nicotinamide adenine dinucleotide is an enteric inhibitory neurotransmitter in human and nonhuman primate colons.Gastroenterology. 2011 Feb;140(2):608-617.e6. doi: 10.1053/j.gastro.2010.09.039. Epub 2010 Sep 25. Gastroenterology. 2011. PMID: 20875415 Free PMC article.
-
P2Y1 purinoreceptors are fundamental to inhibitory motor control of murine colonic excitability and transit.J Physiol. 2012 Apr 15;590(8):1957-72. doi: 10.1113/jphysiol.2011.224634. Epub 2012 Feb 27. J Physiol. 2012. PMID: 22371476 Free PMC article.
-
Platelet-derived growth factor receptor-α-positive cells and not smooth muscle cells mediate purinergic hyperpolarization in murine colonic muscles.Am J Physiol Cell Physiol. 2014 Sep 15;307(6):C561-70. doi: 10.1152/ajpcell.00080.2014. Epub 2014 Jul 23. Am J Physiol Cell Physiol. 2014. PMID: 25055825 Free PMC article.
-
The purinergic neurotransmitter revisited: a single substance or multiple players?Pharmacol Ther. 2014 Nov;144(2):162-91. doi: 10.1016/j.pharmthera.2014.05.012. Epub 2014 Jun 2. Pharmacol Ther. 2014. PMID: 24887688 Free PMC article. Review.
-
Neuronal and extraneuronal release of ATP and NAD(+) in smooth muscle.IUBMB Life. 2012 Oct;64(10):817-24. doi: 10.1002/iub.1076. Epub 2012 Sep 3. IUBMB Life. 2012. PMID: 22941916 Free PMC article. Review.
Cited by
-
Comparison of inhibitory neuromuscular transmission in the Cynomolgus monkey IAS and rectum: special emphasis on differences in purinergic transmission.J Physiol. 2018 Nov;596(22):5319-5341. doi: 10.1113/JP275437. Epub 2018 Oct 13. J Physiol. 2018. PMID: 30198065 Free PMC article.
-
The Imperative for Innovative Enteric Nervous System-Intestinal Organoid Co-Culture Models: Transforming GI Disease Modeling and Treatment.Cells. 2024 May 10;13(10):820. doi: 10.3390/cells13100820. Cells. 2024. PMID: 38786042 Free PMC article. Review.
-
Progress in understanding of inhibitory purinergic neuromuscular transmission in the gut.Neurogastroenterol Motil. 2013 Mar;25(3):203-7. doi: 10.1111/nmo.12090. Neurogastroenterol Motil. 2013. PMID: 23414428 Free PMC article. Review.
-
Urothelial purine release during filling of murine and primate bladders.Am J Physiol Renal Physiol. 2016 Oct 1;311(4):F708-F716. doi: 10.1152/ajprenal.00387.2016. Epub 2016 Jul 27. Am J Physiol Renal Physiol. 2016. PMID: 27465992 Free PMC article.
-
Differential release of β-NAD(+) and ATP upon activation of enteric motor neurons in primate and murine colons.Neurogastroenterol Motil. 2013 Mar;25(3):e194-204. doi: 10.1111/nmo.12069. Epub 2013 Jan 2. Neurogastroenterol Motil. 2013. PMID: 23279315 Free PMC article.
References
-
- Abbracchio MP, Burnstock G, Verkhratsky A, Zimmermann H. Purinergic signalling in the nervous system: an overview. Trends Neurosci. 2009;32:19–29. - PubMed
-
- Agteresch HJ, Dagnelie PC, van den Berg JW, Wilson JH. Adenosine triphosphate: established and potential clinical applications. Drugs. 1999;58:211–232. - PubMed
-
- Bobalova J, Mutafova-Yambolieva VN. Co-release of endogenous ATP and noradrenaline from guinea-pig mesenteric veins exceeds co-release from mesenteric arteries. Clin Exp Pharmacol Physiol. 2001;28:397–401. - PubMed
-
- Bobalova J, Mutafova-Yambolieva VN. Membrane-bound and releasable nucleotidase activities: differences in canine mesenteric artery and vein. Clin Exp Pharmacol Physiol. 2003;30:194–202. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials