

Epidermal growth factor chronically upregulates Ca(2+)-dependent Cl(-) conductance and TMEM16A expression in intestinal epithelial cells
- PMID: 22351639
- PMCID: PMC3573312
- DOI: 10.1113/jphysiol.2011.226126
Epidermal growth factor chronically upregulates Ca(2+)-dependent Cl(-) conductance and TMEM16A expression in intestinal epithelial cells
Abstract
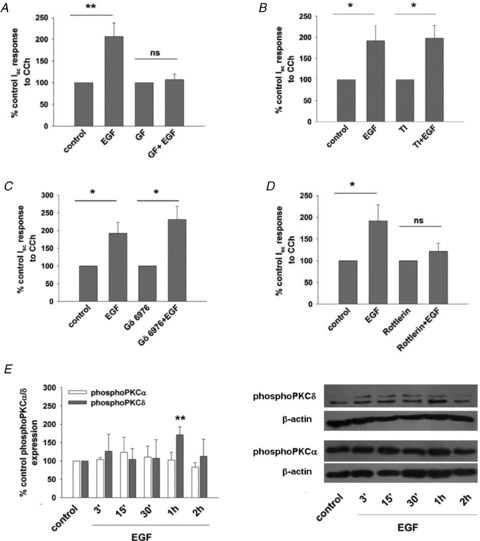
Dysregulated epithelial fluid and electrolyte transport is a common feature of many intestinal disorders. However, molecular mechanisms that regulate epithelial transport processes are still poorly understood, thereby limiting development of new therapeutics. Previously, we showed that epidermal growth factor (EGF) chronically enhances intestinal epithelial secretory function. Here, we investigated a potential role for altered expression or activity of apical Cl(−) channels in mediating the effects of EGF. Cl(−) secretion across monolayers of T(84) colonic epithelia was measured as changes in short-circuit current. Protein expression/phosphorylation was measured by RT-PCR and Western blotting. Under conditions that specifically isolate apical Ca(2+)-activated Cl(−) channel (CaCC) currents, EGF pretreatment (100 ng ml(−1) for 15 min) potentiated carbachol (CCh)-induced responses to 173 ± 25% of those in control cells, when measured 24 h later (n = 26; P < 0.01). EGF-induced increases in CaCC currents were abolished by the transmembrane protein 16A (TMEM16A) inhibitor, T16A(inh)-A01 (10 μm). Furthermore, TMEM16A mRNA and protein expression was increased by EGF to 256 ± 38% (n = 7; P < 0.01) and 297 ± 46% (n = 9, P < 0.001) of control levels, respectively. In contrast, EGF did not alter CFTR expression or activity. EGF-induced increases in Cl(−) secretion, CaCC currents and TMEM16A expression were attenuated by a PKCδ inhibitor, rottlerin (20 μm), and a phosphatidylinositol 3-kinase (PI3K) inhibitor, LY290042 (25 μm). Finally, LY290042 inhibited EGF-induced phosphorylation of PKCδ. We conclude that EGF chronically upregulates Ca(2+)-dependent Cl(−) conductances and TMEM16A expression in intestinal epithelia by a mechanism involving sequential activation of PI3K and PKCδ. Therapeutic targeting of EGF receptor-dependent signalling pathways may provide new approaches for treatment of epithelial transport disorders.
Figures




Similar articles
-
TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells.J Biol Chem. 2011 Jan 21;286(3):2365-74. doi: 10.1074/jbc.M110.175109. Epub 2010 Nov 17. J Biol Chem. 2011. PMID: 21084298 Free PMC article.
-
Induction of Na+/K+/2Cl- cotransporter expression mediates chronic potentiation of intestinal epithelial Cl- secretion by EGF.Am J Physiol Cell Physiol. 2008 Jun;294(6):C1362-70. doi: 10.1152/ajpcell.00256.2007. Epub 2008 Apr 9. Am J Physiol Cell Physiol. 2008. PMID: 18400987
-
Dextran sulfate sodium-induced chronic colitis attenuates Ca2+-activated Cl- secretion in murine colon by downregulating TMEM16A.Am J Physiol Cell Physiol. 2018 Jul 1;315(1):C10-C20. doi: 10.1152/ajpcell.00328.2017. Epub 2018 Mar 21. Am J Physiol Cell Physiol. 2018. PMID: 29561662 Free PMC article.
-
Bestrophin and TMEM16-Ca(2+) activated Cl(-) channels with different functions.Cell Calcium. 2009 Oct;46(4):233-41. doi: 10.1016/j.ceca.2009.09.003. Epub 2009 Sep 26. Cell Calcium. 2009. PMID: 19783045 Review.
-
Anoctamin 1 in secretory epithelia.Cell Calcium. 2014 Jun;55(6):355-61. doi: 10.1016/j.ceca.2014.02.006. Epub 2014 Feb 15. Cell Calcium. 2014. PMID: 24636668 Review.
Cited by
-
Potentiation of calcium-activated chloride secretion and barrier dysfunction may underlie EGF receptor tyrosine kinase inhibitor-induced diarrhea.Physiol Rep. 2020 Jul;8(13):e14490. doi: 10.14814/phy2.14490. Physiol Rep. 2020. PMID: 32652816 Free PMC article.
-
Role of Epidermal Growth Factor Receptor (EGFR) and Its Ligands in Kidney Inflammation and Damage.Mediators Inflamm. 2018 Dec 23;2018:8739473. doi: 10.1155/2018/8739473. eCollection 2018. Mediators Inflamm. 2018. PMID: 30670929 Free PMC article. Review.
-
Evidence for metabotropic function of epithelial nicotinic cholinergic receptors in rat colon.Br J Pharmacol. 2019 May;176(9):1328-1340. doi: 10.1111/bph.14638. Epub 2019 Apr 9. Br J Pharmacol. 2019. PMID: 30807644 Free PMC article.
-
Epidermal growth factor, from gene organization to bedside.Semin Cell Dev Biol. 2014 Apr;28:2-11. doi: 10.1016/j.semcdb.2014.01.011. Epub 2014 Feb 7. Semin Cell Dev Biol. 2014. PMID: 24513230 Free PMC article. Review.
-
The multifaceted role of TMEM16A in cancer.Cell Calcium. 2019 Sep;82:102050. doi: 10.1016/j.ceca.2019.06.004. Epub 2019 Jun 14. Cell Calcium. 2019. PMID: 31279157 Free PMC article. Review.
References
-
- Barrett KE, Keely SJ. Chloride secretion by the intestinal epithelium: molecular basis and regulatory aspects. Annu Rev Physiol. 2000;62:535–572. - PubMed
-
- Berschneider HM, Knowles MR, Azizkhan RG, Boucher RC, Tobey NA, Orlando RC, Powell DW. Altered intestinal chloride transport in cystic fibrosis. FASEB J. 1988;2:2625–2629. - PubMed
-
- Bertelsen LS, Barrett KE, Keely SJ. Gs protein-coupled receptor agonists induce transactivation of the epidermal growth factor receptor in T84 cells: implications for epithelial secretory responses. J Biol Chem. 2004;279:6271–6279. - PubMed
-
- Borok Z, Hami A, Danto SI, Lubman RL, Kim KJ, Crandall ED. Effects of EGF on alveolar epithelial junctional permeability and active sodium transport. Am J Physiol Lung Cell Mol Physiol. 1996;270:L559–565. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous