

Toward a unifying hypothesis of metabolic syndrome
- PMID: 22351884
- PMCID: PMC3289531
- DOI: 10.1542/peds.2011-2912
Toward a unifying hypothesis of metabolic syndrome
Abstract
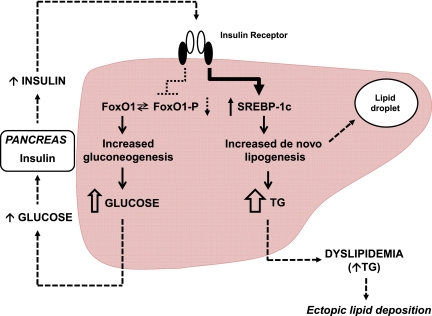
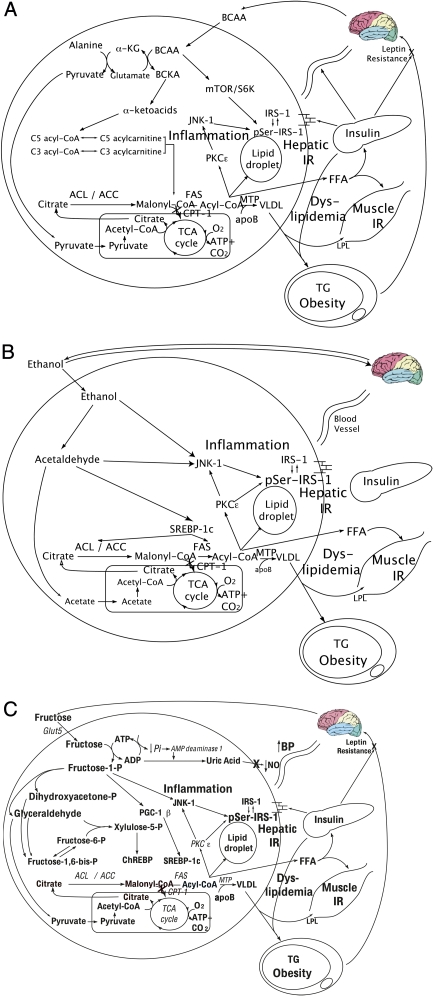
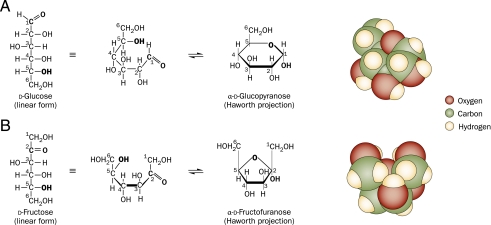
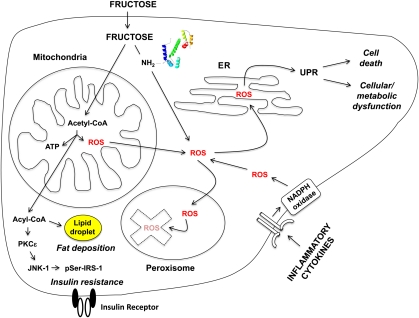
Despite a lack of consistent diagnostic criteria, the metabolic syndrome (MetS) is increasingly evident in children and adolescents, portending a tsunami of chronic disease and mortality as this generation ages. The diagnostic criteria for MetS apply absolute cutoffs to continuous variables and fail to take into account aging, pubertal changes, and race/ethnicity. We attempt to define MetS mechanistically to determine its specific etiologies and to identify targets for therapy. Whereas the majority of studies document a relationship of visceral fat to insulin resistance, ectopic liver fat correlates better with dysfunctional insulin dynamics from which the rest of MetS derives. In contrast to the systemic metabolism of glucose, the liver is the primary metabolic clearinghouse for 4 specific foodstuffs that have been associated with the development of MetS: trans-fats, branched-chain amino acids, ethanol, and fructose. These 4 substrates (1) are not insulin regulated and (2) deliver metabolic intermediates to hepatic mitochondria without an appropriate "pop-off" mechanism for excess substrate, enhancing lipogenesis and ectopic adipose storage. Excessive fatty acid derivatives interfere with hepatic insulin signal transduction. Reactive oxygen species accumulate, which cannot be quenched by adjacent peroxisomes; these reactive oxygen species reach the endoplasmic reticulum, leading to a compensatory process termed the "unfolded protein response," driving further insulin resistance and eventually insulin deficiency. No obvious drug target exists in this pathway; thus, the only rational therapeutic approaches remain (1) altering hepatic substrate availability (dietary modification), (2) reducing hepatic substrate flux (high fiber), or (3) increasing mitochondrial efficiency (exercise).
Figures
References
-
- Chan JM, Rimm EB, Colditz GA, Stampfer MJ, Willett WC. Obesity, fat distribution, and weight gain as risk factors for clinical diabetes in men. Diabetes Care. 1994;17(9):961–969 - PubMed
-
- Wildman RP. Healthy obesity. Curr Opin Clin Nutr Metab Care. 2009;12(4):438–443 - PubMed
-
- Conus F, Rabasa-Lhoret R, Péronnet F. Characteristics of metabolically obese normal-weight (MONW) subjects. Appl Physiol Nutr Metab. 2007;32(1):4–12 - PubMed
-
- Ruderman N, Chisholm D, Pi-Sunyer X, Schneider S. The metabolically obese, normal-weight individual revisited. Diabetes. 1998;47(5):699–713 - PubMed
-
- Abbasi F, Brown BW, Jr, Lamendola C, McLaughlin T, Reaven GM. Relationship between obesity, insulin resistance, and coronary heart disease risk. J Am Coll Cardiol. 2002;40(5):937–943 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
