

Ovarian cancer progression is controlled by phenotypic changes in dendritic cells
- PMID: 22351930
- PMCID: PMC3302234
- DOI: 10.1084/jem.20111413
Ovarian cancer progression is controlled by phenotypic changes in dendritic cells
Abstract
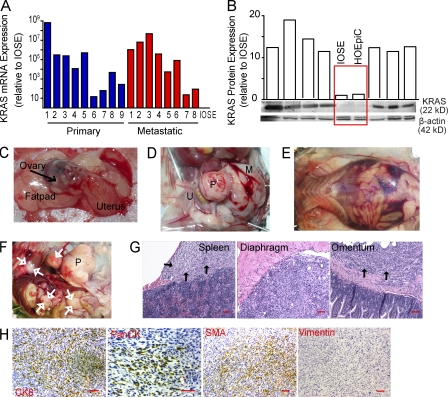
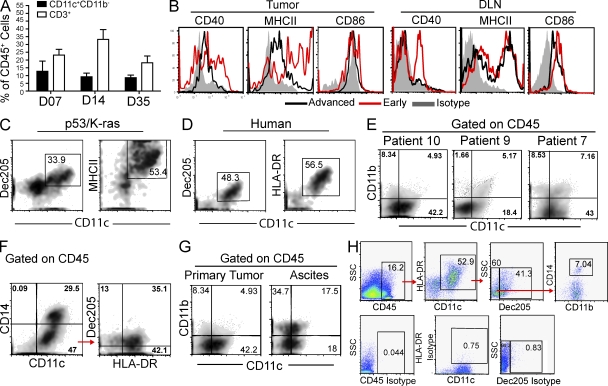
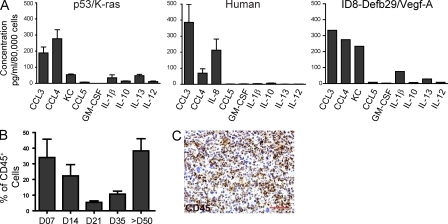
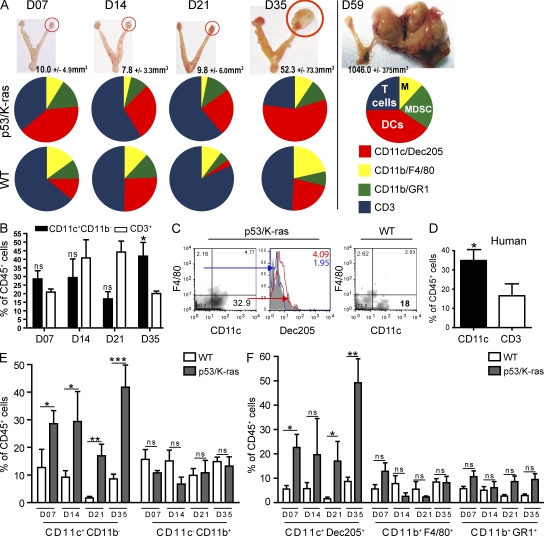
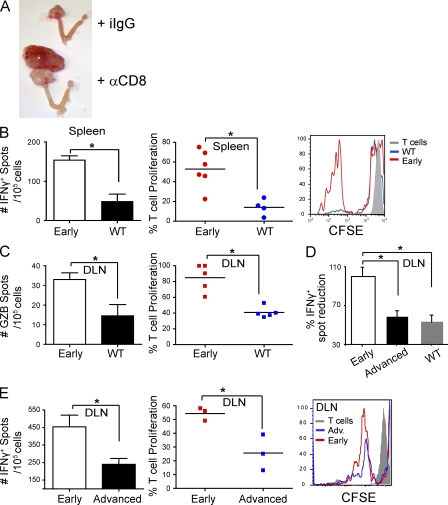
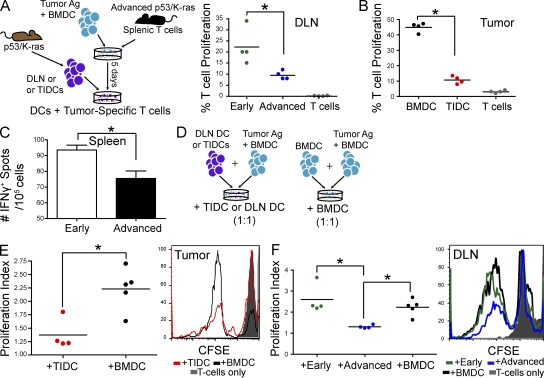
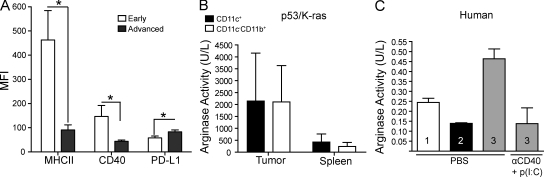
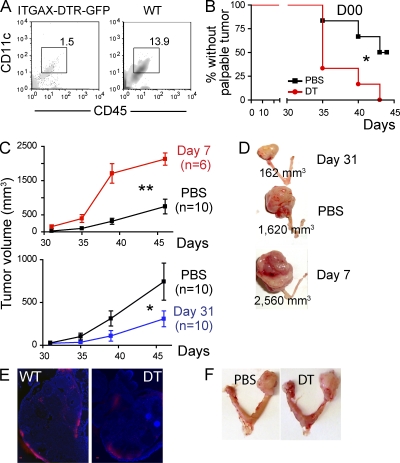
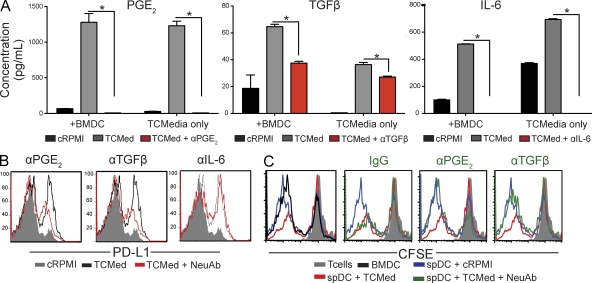
We characterized the initiation and evolution of the immune response against a new inducible p53-dependent model of aggressive ovarian carcinoma that recapitulates the leukocyte infiltrates and cytokine milieu of advanced human tumors. Unlike other models that initiate tumors before the development of a mature immune system, we detect measurable anti-tumor immunity from very early stages, which is driven by infiltrating dendritic cells (DCs) and prevents steady tumor growth for prolonged periods. Coinciding with a phenotypic switch in expanding DC infiltrates, tumors aggressively progress to terminal disease in a comparatively short time. Notably, tumor cells remain immunogenic at advanced stages, but anti-tumor T cells become less responsive, whereas their enduring activity is abrogated by different microenvironmental immunosuppressive DCs. Correspondingly, depleting DCs early in the disease course accelerates tumor expansion, but DC depletion at advanced stages significantly delays aggressive malignant progression. Our results indicate that phenotypically divergent DCs drive both immunosurveillance and accelerated malignant growth. We provide experimental support for the cancer immunoediting hypothesis, but we also show that aggressive cancer progression after a comparatively long latency period is primarily driven by the mobilization of immunosuppressive microenvironmental leukocytes, rather than loss of tumor immunogenicity.
Figures
Comment in
-
Tumour immunology: dendritic cell switch.Nat Rev Cancer. 2012 Mar 15;12(4):230. doi: 10.1038/nrc3253. Nat Rev Cancer. 2012. PMID: 22419255 No abstract available.
References
-
- Connolly D.C., Bao R., Nikitin A.Y., Stephens K.C., Poole T.W., Hua X., Harris S.S., Vanderhyden B.C., Hamilton T.C. 2003. Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer. Cancer Res. 63:1389–1397 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P30CA10815/CA/NCI NIH HHS/United States
- U54 CA151662/CA/NCI NIH HHS/United States
- R01CA157664/CA/NCI NIH HHS/United States
- F31 CA134188/CA/NCI NIH HHS/United States
- P30 CA010815/CA/NCI NIH HHS/United States
- R01CA124515/CA/NCI NIH HHS/United States
- U54CA151662/CA/NCI NIH HHS/United States
- R21CA132026/CA/NCI NIH HHS/United States
- R01 CA124515/CA/NCI NIH HHS/United States
- F31CA134188/CA/NCI NIH HHS/United States
- R01CA124515S/CA/NCI NIH HHS/United States
- R01 CA157664/CA/NCI NIH HHS/United States
- P30 CA023108/CA/NCI NIH HHS/United States
- R21 CA132026/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
