

JNK2 and JNK3 are major regulators of axonal injury-induced retinal ganglion cell death
- PMID: 22353563
- PMCID: PMC3323666
- DOI: 10.1016/j.nbd.2012.02.003
JNK2 and JNK3 are major regulators of axonal injury-induced retinal ganglion cell death
Abstract
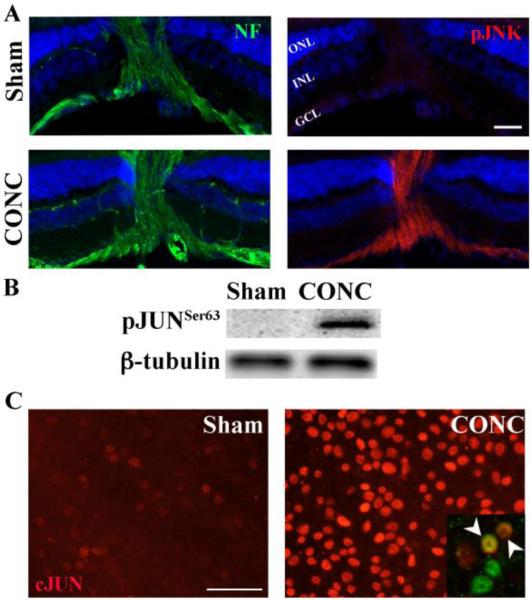
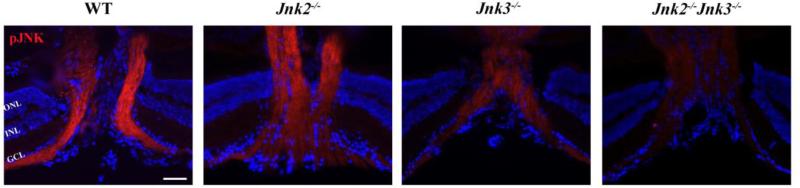
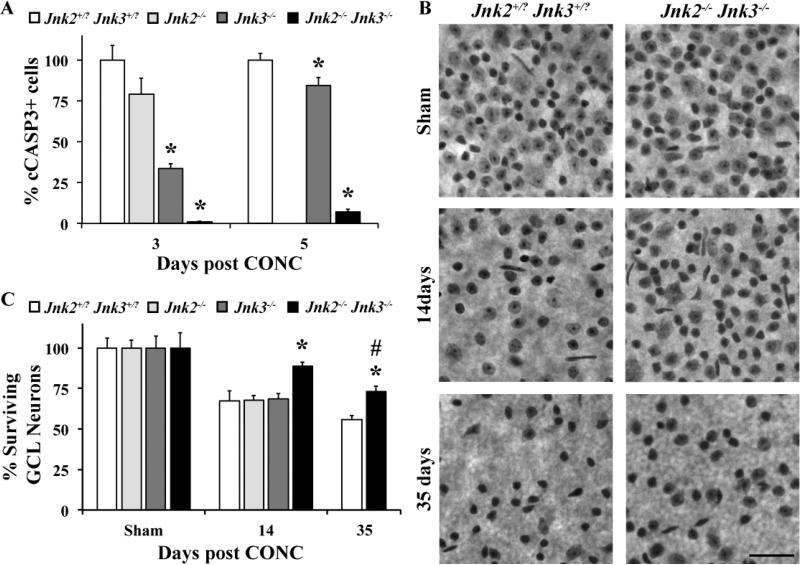
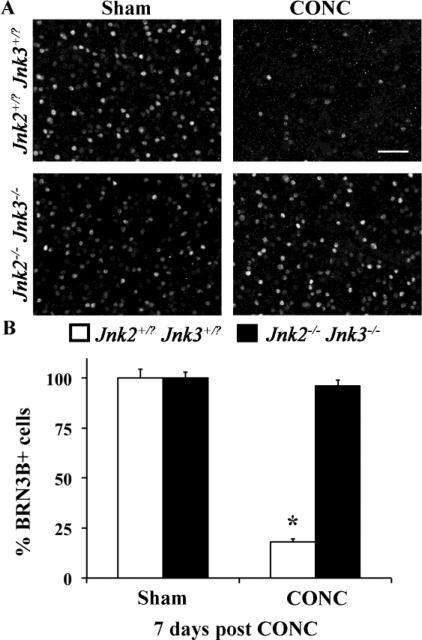
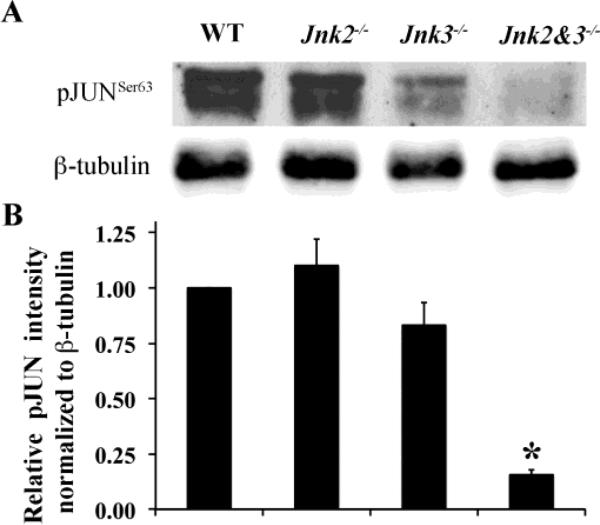
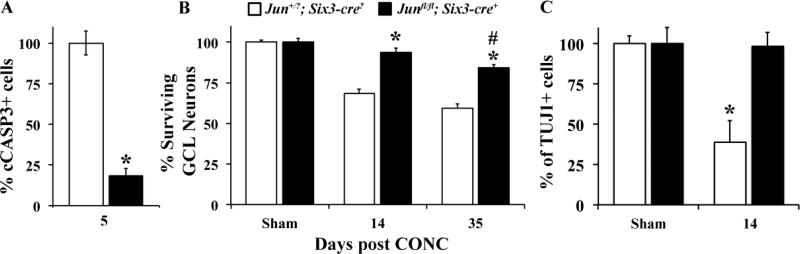
Glaucoma is a neurodegenerative disease characterized by the apoptotic death of retinal ganglion cells (RGCs). The primary insult to RGCs in glaucoma is thought to occur to their axons as they exit the eye in the optic nerve head. However, pathological signaling pathways that exert central roles in triggering RGC death following axonal injury remain unidentified. It is likely that the first changes to occur following axonal injury are signal relay events that transduce the injury signal from the axon to the cell body. Here we focus on the c-Jun N-terminal kinase (JNK1-3) family, a signaling pathway implicated in axonal injury signaling and neurodegenerative apoptosis, and likely to function as a central node in axonal injury-induced RGC death. We show that JNK signaling is activated immediately after axonal injury in RGC axons at the site of injury. Following its early activation, sustained JNK signaling is observed in axonally-injured RGCs in the form of JUN phosphorylation and upregulation. Using mice lacking specific Jnk isoforms, we show that Jnk2 and Jnk3 are the isoforms activated in injured axons. Combined deficiency of Jnk2 and Jnk3 provides robust long-term protection against axonal injury-induced RGC death and prevents downregulation of the RGC marker, BRN3B, and phosphorylation of JUN. Finally, using Jun deficient mice, we show that JUN-dependent pathways are important for axonal injury-induced RGC death. Together these data demonstrate that JNK signaling is the major early pathway triggering RGC death after axonal injury and may directly link axon injury to transcriptional activity that controls RGC death.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
