

Pathways for repairing and tolerating the spectrum of oxidative DNA lesions
- PMID: 22353689
- PMCID: PMC3389563
- DOI: 10.1016/j.canlet.2012.02.001
Pathways for repairing and tolerating the spectrum of oxidative DNA lesions
Abstract
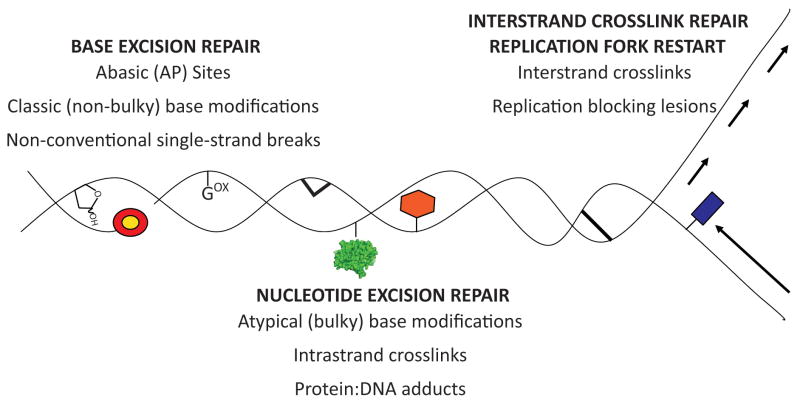
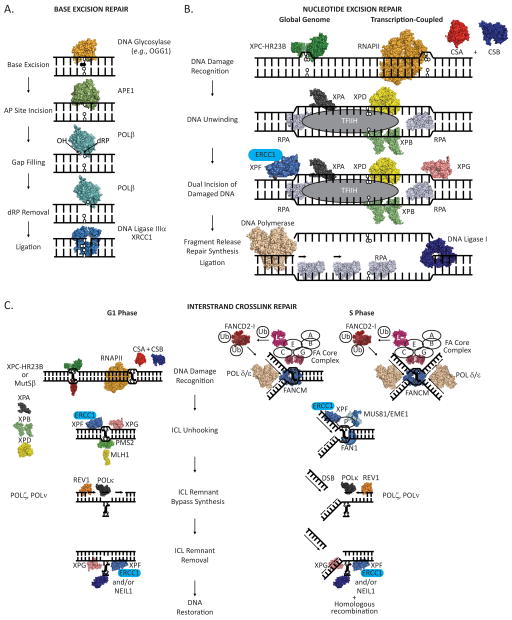
Reactive oxygen species (ROS) arise from both endogenous and exogenous sources. These reactive molecules possess the ability to damage both the DNA nucleobases and the sugar phosphate backbone, leading to a wide spectrum of lesions, including non-bulky (8-oxoguanine and formamidopyrimidine) and bulky (cyclopurine and etheno adducts) base modifications, abasic sites, non-conventional single-strand breaks, protein-DNA adducts, and intra/interstrand DNA crosslinks. Unrepaired oxidative DNA damage can result in bypass mutagenesis during genome copying or gene expression, or blockage of the essential cellular processes of DNA replication or transcription. Such outcomes underlie numerous pathologies, including, but not limited to, carcinogenesis and neurodegeneration, as well as the aging process. Cells have adapted and evolved defense systems against the deleterious effects of ROS, and specifically devote a number of cellular DNA repair and tolerance pathways to combat oxidative DNA damage. Defects in these protective pathways trigger hereditary human diseases that exhibit increased cancer incidence, developmental defects, neurological abnormalities, and/or premature aging. We review herein classic and atypical oxidative DNA lesions, outcomes of encountering these damages during DNA replication and transcription, and the consequences of losing the ability to repair the different forms of oxidative DNA damage. We particularly focus on the hereditary human diseases Xeroderma Pigmentosum, Cockayne Syndrome and Fanconi Anemia, which may involve defects in the efficient repair of oxidative modifications to chromosomal DNA.
Published by Elsevier Ireland Ltd.
Figures
References
-
- Imlay JA, Linn S. DNA damage and oxygen radical toxicity. Science. 1988;240:1302–1309. - PubMed
-
- Mannaerts GP, Van Veldhoven PP. Metabolic pathways in mammalian peroxisomes. Biochimie. 1993;75:147–158. - PubMed
-
- Shen Z, Wu W, Hazen SL. Activated leukocytes oxidatively damage DNA, RNA, and the nucleotide pool through halide-dependent formation of hydroxyl radical. Biochemistry. 2000;39:5474–5482. - PubMed
-
- Kovacic P, Wakelin LP. Review: DNA molecular electrostatic potential: novel perspectives for the mechanism of action of anticancer drugs involving electron transfer and oxidative stress. Anticancer Drug Des. 2001;16:175–184. - PubMed
-
- Wood ML, Dizdaroglu M, Gajewski E, Essigmann JM. Mechanistic studies of ionizing radiation and oxidative mutagenesis: genetic effects of a single 8-hydroxyguanine (7-hydro-8-oxoguanine) residue inserted at a unique site in a viral genome. Biochemistry. 1990;29:7024–7032. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
