

Hypervariable loci in the human gut virome
- PMID: 22355105
- PMCID: PMC3309749
- DOI: 10.1073/pnas.1119061109
Hypervariable loci in the human gut virome
Abstract
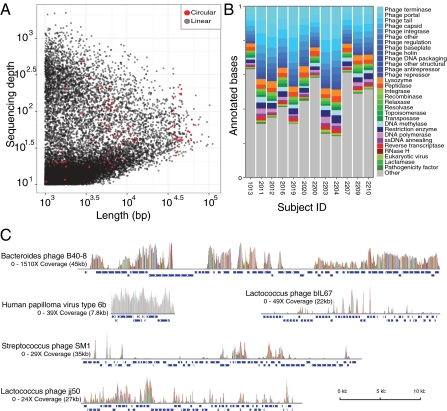
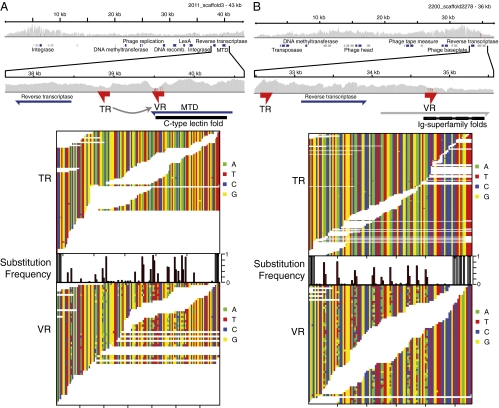
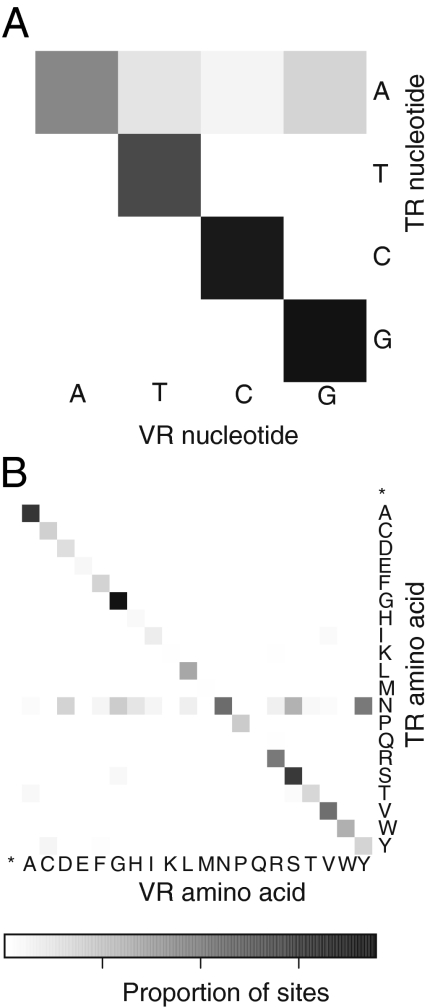
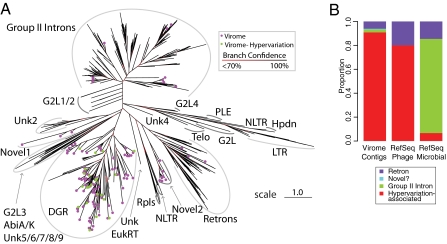
Genetic variation is critical in microbial immune evasion and drug resistance, but variation has rarely been studied in complex heterogeneous communities such as the human microbiome. To begin to study natural variation, we analyzed DNA viruses present in the lower gastrointestinal tract of 12 human volunteers by determining 48 billion bases of viral DNA sequence. Viral genomes mostly showed low variation, but 51 loci of ∼100 bp showed extremely high variation, so that up to 96% of the viral genomes encoded unique amino acid sequences. Some hotspots of hypervariation were in genes homologous to the bacteriophage BPP-1 viral tail-fiber gene, which is known to be hypermutagenized by a unique reverse-transcriptase (RT)-based mechanism. Unexpectedly, other hypervariable loci in our data were in previously undescribed gene types, including genes encoding predicted Ig-superfamily proteins. Most of the hypervariable loci were linked to genes encoding RTs of a single clade, which we find is the most abundant clade among gut viruses but only a minor component of bacterial RT populations. Hypervariation was targeted to 5'-AAY-3' asparagine codons, which allows maximal chemical diversification of the encoded amino acids while avoiding formation of stop codons. These findings document widespread targeted hypervariation in the human gut virome, identify previously undescribed types of genes targeted for hypervariation, clarify association with RT gene clades, and motivate studies of hypervariation in the full human microbiome.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Craig NL, Craigie R, Gellert M, Lambowitz AM. Mobile DNA II. Washington, DC: ASM,; 2002.
-
- Bushman FD. Lateral DNA Transfer: Mechanisms and Consequences. Cold Spring Harbor, NY: Cold Spring Harbor Lab Press; 2001.
-
- McMahon SA, et al. The C-type lectin fold as an evolutionary solution for massive sequence variation. Nat Struct Mol Biol. 2005;12:886–892. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
