

An evolutionary network of genes present in the eukaryote common ancestor polls genomes on eukaryotic and mitochondrial origin
- PMID: 22355196
- PMCID: PMC3342870
- DOI: 10.1093/gbe/evs018
An evolutionary network of genes present in the eukaryote common ancestor polls genomes on eukaryotic and mitochondrial origin
Abstract
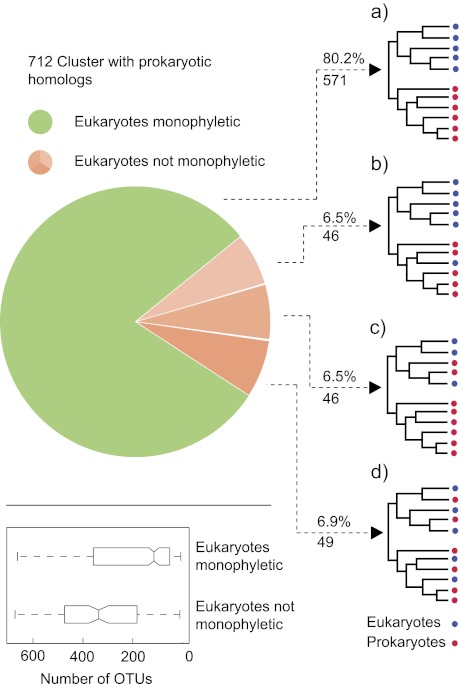
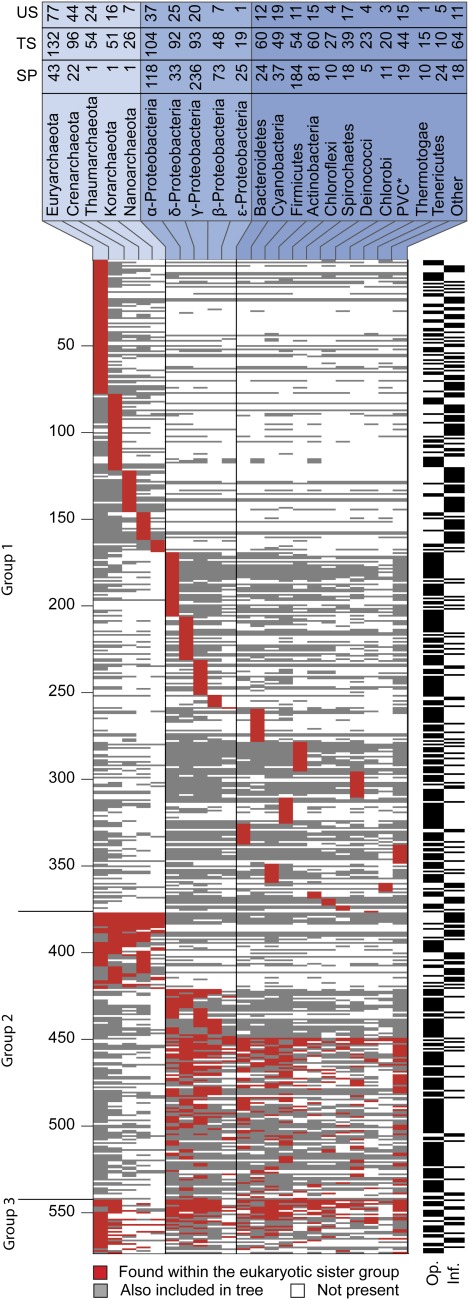
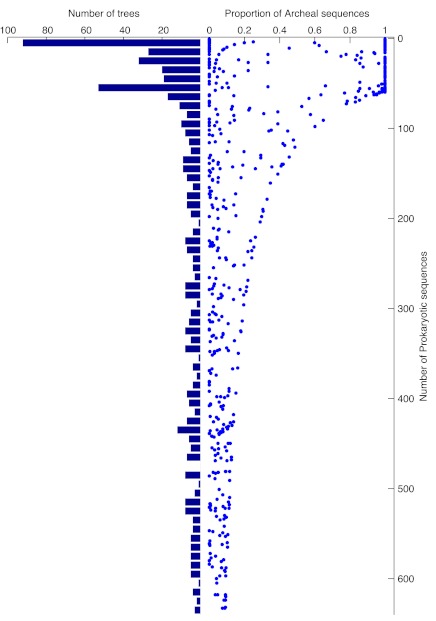
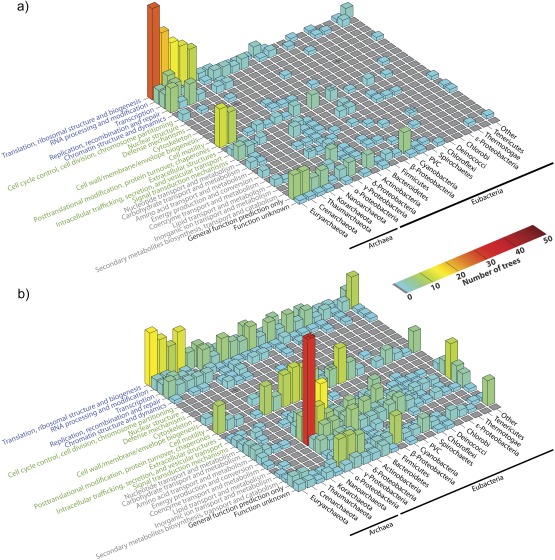
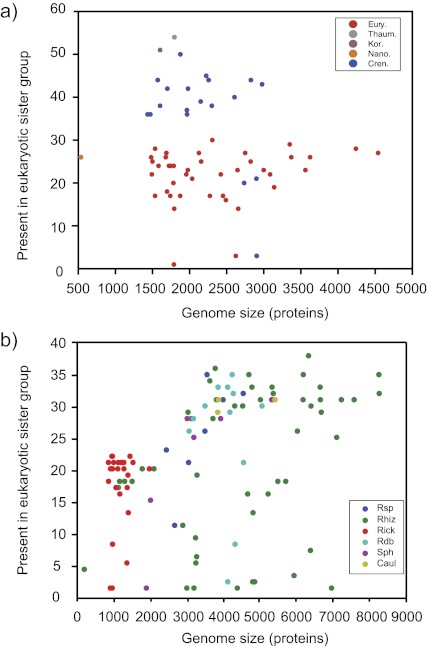
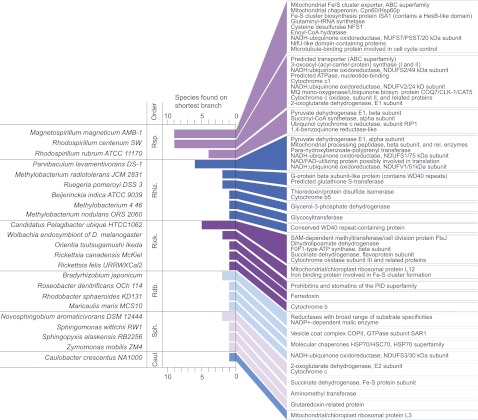
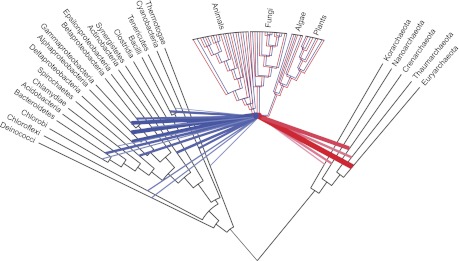
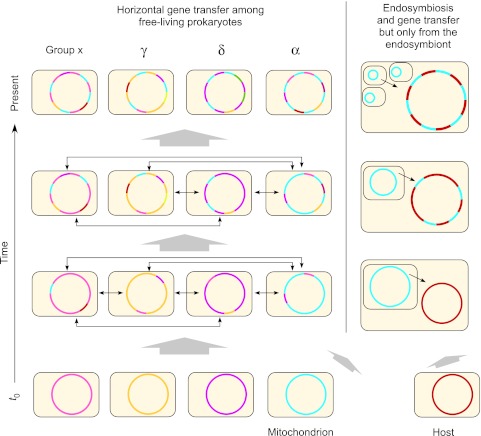
To test the predictions of competing and mutually exclusive hypotheses for the origin of eukaryotes, we identified from a sample of 27 sequenced eukaryotic and 994 sequenced prokaryotic genomes 571 genes that were present in the eukaryote common ancestor and that have homologues among eubacterial and archaebacterial genomes. Maximum-likelihood trees identified the prokaryotic genomes that most frequently contained genes branching as the sister to the eukaryotic nuclear homologues. Among the archaebacteria, euryarchaeote genomes most frequently harbored the sister to the eukaryotic nuclear gene, whereas among eubacteria, the α-proteobacteria were most frequently represented within the sister group. Only 3 genes out of 571 gave a 3-domain tree. Homologues from α-proteobacterial genomes that branched as the sister to nuclear genes were found more frequently in genomes of facultatively anaerobic members of the rhiozobiales and rhodospirilliales than in obligate intracellular ricketttsial parasites. Following α-proteobacteria, the most frequent eubacterial sister lineages were γ-proteobacteria, δ-proteobacteria, and firmicutes, which were also the prokaryote genomes least frequently found as monophyletic groups in our trees. Although all 22 higher prokaryotic taxa sampled (crenarchaeotes, γ-proteobacteria, spirochaetes, chlamydias, etc.) harbor genes that branch as the sister to homologues present in the eukaryotic common ancestor, that is not evidence of 22 different prokaryotic cells participating at eukaryote origins because prokaryotic "lineages" have laterally acquired genes for more than 1.5 billion years since eukaryote origins. The data underscore the archaebacterial (host) nature of the eukaryotic informational genes and the eubacterial (mitochondrial) nature of eukaryotic energy metabolism. The network linking genes of the eukaryote ancestor to contemporary homologues distributed across prokaryotic genomes elucidates eukaryote gene origins in a dialect cognizant of gene transfer in nature.
Figures

References
-
- Abhishek A, Bavishi A, Choudhary M. Bacterial genome chimaerism and the origin of mitochondria. Can J Microbiol. 2011;57:49–61. - PubMed
-
- Atteia A, et al. Pyruvate formate-lyase and a novel route of eukaryotic ATP-synthesis in anaerobic Chlamydomonas mitochondria. J Biol Chem. 2006;281:9909–9918. - PubMed
-
- Atteia A, et al. A proteomic survey of Chlamydomonas reinhardtii mitochondria sheds new light on the metabolic plasticity of the organelle and on the nature of the α-proteobacterial mitochondrial ancestor. Mol Biol Evol. 2009;29:1533–1548. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
