

Charged and hydrophobic surfaces on the a chain of shiga-like toxin 1 recognize the C-terminal domain of ribosomal stalk proteins
- PMID: 22355345
- PMCID: PMC3280276
- DOI: 10.1371/journal.pone.0031191
Charged and hydrophobic surfaces on the a chain of shiga-like toxin 1 recognize the C-terminal domain of ribosomal stalk proteins
Abstract
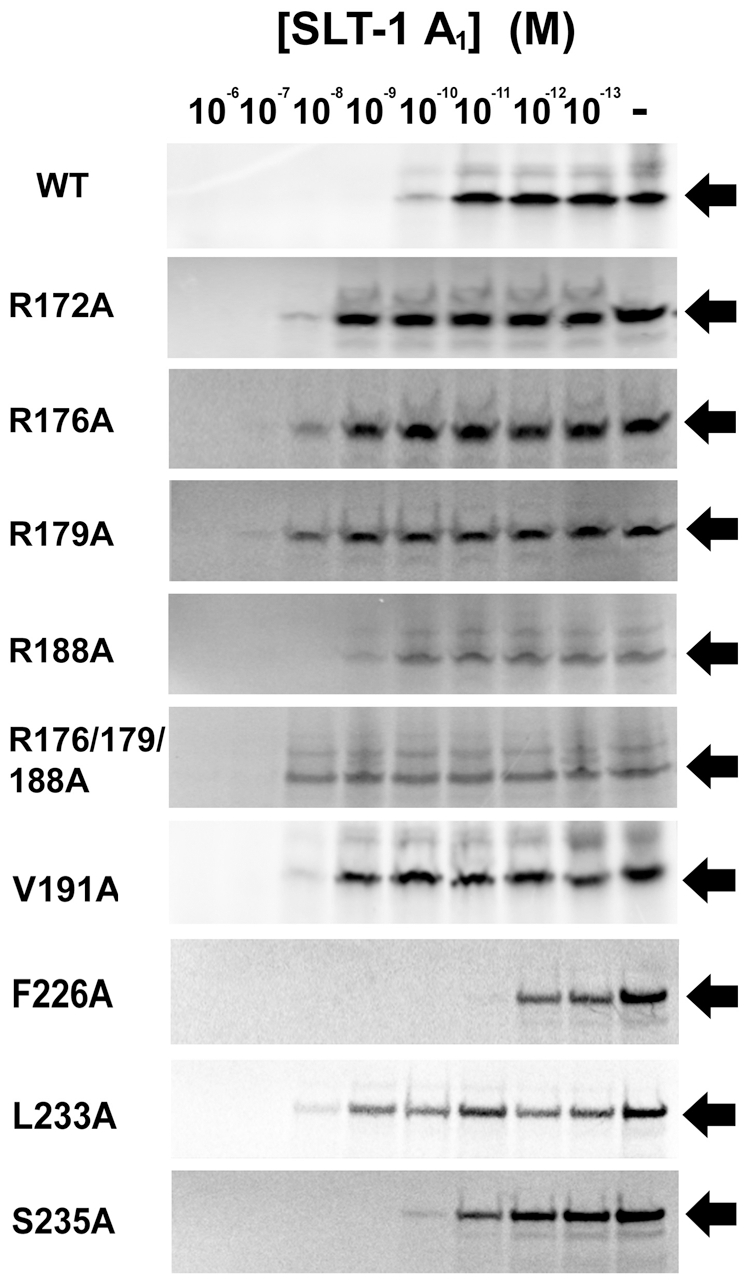
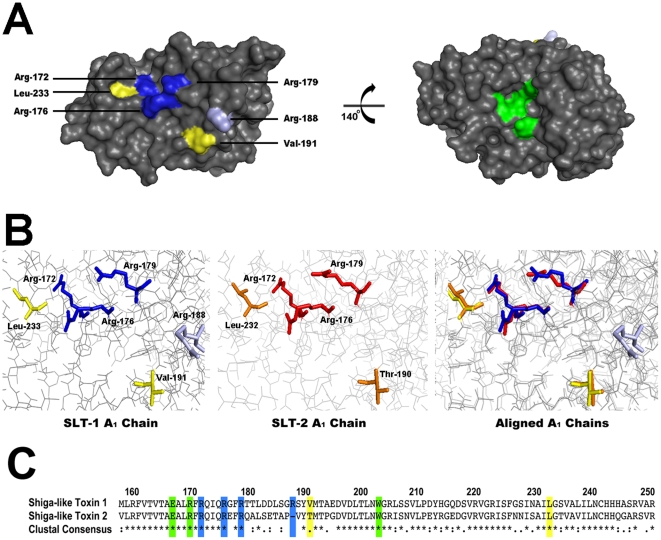
Shiga-like toxins are ribosome-inactivating proteins (RIP) produced by pathogenic E. coli strains that are responsible for hemorrhagic colitis and hemolytic uremic syndrome. The catalytic A(1) chain of Shiga-like toxin 1 (SLT-1), a representative RIP, first docks onto a conserved peptide SD[D/E]DMGFGLFD located at the C-terminus of all three eukaryotic ribosomal stalk proteins and halts protein synthesis through the depurination of an adenine base in the sarcin-ricin loop of 28S rRNA. Here, we report that the A(1) chain of SLT-1 rapidly binds to and dissociates from the C-terminal peptide with a monomeric dissociation constant of 13 µM. An alanine scan performed on the conserved peptide revealed that the SLT-1 A(1) chain interacts with the anionic tripeptide DDD and the hydrophobic tetrapeptide motif FGLF within its sequence. Based on these 2 peptide motifs, SLT-1 A(1) variants were generated that displayed decreased affinities for the stalk protein C-terminus and also correlated with reduced ribosome-inactivating activities in relation to the wild-type A(1) chain. The toxin-peptide interaction and subsequent toxicity were shown to be mediated by cationic and hydrophobic docking surfaces on the SLT-1 catalytic domain. These docking surfaces are located on the opposite face of the catalytic cleft and suggest that the docking of the A(1) chain to SDDDMGFGLFD may reorient its catalytic domain to face its RNA substrate. More importantly, both the delineated A(1) chain ribosomal docking surfaces and the ribosomal peptide itself represent a target and a scaffold, respectively, for the design of generic inhibitors to block the action of RIPs.
Conflict of interest statement
Figures
Similar articles
-
The catalytic subunit of shiga-like toxin 1 interacts with ribosomal stalk proteins and is inhibited by their conserved C-terminal domain.J Mol Biol. 2008 Apr 25;378(2):375-86. doi: 10.1016/j.jmb.2008.02.014. Epub 2008 Feb 15. J Mol Biol. 2008. PMID: 18358491
-
Structural basis for the interaction of Shiga toxin 2a with a C-terminal peptide of ribosomal P stalk proteins.J Biol Chem. 2020 Nov 13;295(46):15588-15596. doi: 10.1074/jbc.AC120.015070. Epub 2020 Sep 2. J Biol Chem. 2020. PMID: 32878986 Free PMC article.
-
The ribosomal stalk is required for ribosome binding, depurination of the rRNA and cytotoxicity of ricin A chain in Saccharomyces cerevisiae.Mol Microbiol. 2008 Dec;70(6):1441-52. doi: 10.1111/j.1365-2958.2008.06492.x. Epub 2008 Oct 30. Mol Microbiol. 2008. PMID: 19019145 Free PMC article.
-
Interaction of ricin and Shiga toxins with ribosomes.Curr Top Microbiol Immunol. 2012;357:1-18. doi: 10.1007/82_2011_174. Curr Top Microbiol Immunol. 2012. PMID: 21910078 Free PMC article. Review.
-
Structures and Ribosomal Interaction of Ribosome-Inactivating Proteins.Molecules. 2016 Nov 21;21(11):1588. doi: 10.3390/molecules21111588. Molecules. 2016. PMID: 27879643 Free PMC article. Review.
Cited by
-
Crystal Structure of Ribosome-Inactivating Protein Ricin A Chain in Complex with the C-Terminal Peptide of the Ribosomal Stalk Protein P2.Toxins (Basel). 2016 Oct 13;8(10):296. doi: 10.3390/toxins8100296. Toxins (Basel). 2016. PMID: 27754366 Free PMC article.
-
Targeting ricin to the ribosome.Toxicon. 2013 Jul;69:143-51. doi: 10.1016/j.toxicon.2013.02.001. Epub 2013 Feb 20. Toxicon. 2013. PMID: 23454625 Free PMC article. Review.
-
Facing glycosphingolipid-Shiga toxin interaction: dire straits for endothelial cells of the human vasculature.Cell Mol Life Sci. 2013 Feb;70(3):425-57. doi: 10.1007/s00018-012-1060-z. Epub 2012 Jul 6. Cell Mol Life Sci. 2013. PMID: 22766973 Free PMC article. Review.
-
Ribosomal stalk proteins RPLP1 and RPLP2 promote biogenesis of flaviviral and cellular multi-pass transmembrane proteins.Nucleic Acids Res. 2020 Sep 25;48(17):9872-9885. doi: 10.1093/nar/gkaa717. Nucleic Acids Res. 2020. PMID: 32890404 Free PMC article.
-
Extensive Evolution of Cereal Ribosome-Inactivating Proteins Translates into Unique Structural Features, Activation Mechanisms, and Physiological Roles.Toxins (Basel). 2017 Mar 29;9(4):123. doi: 10.3390/toxins9040123. Toxins (Basel). 2017. PMID: 28353660 Free PMC article. Review.
References
-
- Riley LW. The epidemiologic, clinical, and microbiologic features of hemorrhagic colitis. Annu Rev Microbiol. 1987;41:383–407. - PubMed
-
- Fraser ME, Chernaia MM, Kozlov YV, James MN. Crystal structure of the holotoxin from Shigella dysenteriae at 2.5 A resolution. Nat Struct Biol. 1994;1:59–64. - PubMed
-
- Kozlov YV, Chernaia MM, Fraser ME, James MN. Purification and crystallization of Shiga toxin from Shigella dysenteriae. J Mol Biol. 1993;232:704–706. - PubMed
-
- Lindberg AA, Brown JE, Stromberg N, Westling-Ryd M, Schultz JE, et al. Identification of the carbohydrate receptor for Shiga toxin produced by Shigella dysenteriae type 1. J Biol Chem. 1987;262:1779–1785. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
