

High resolution methylome map of rat indicates role of intragenic DNA methylation in identification of coding region
- PMID: 22355382
- PMCID: PMC3280313
- DOI: 10.1371/journal.pone.0031621
High resolution methylome map of rat indicates role of intragenic DNA methylation in identification of coding region
Abstract
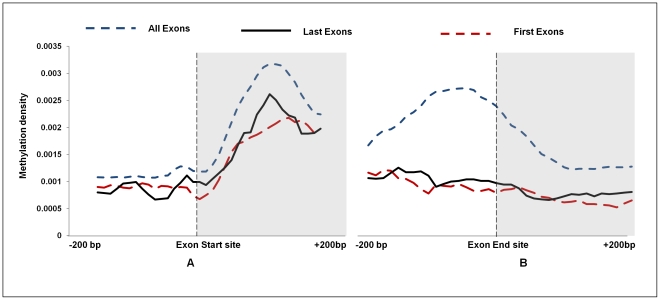
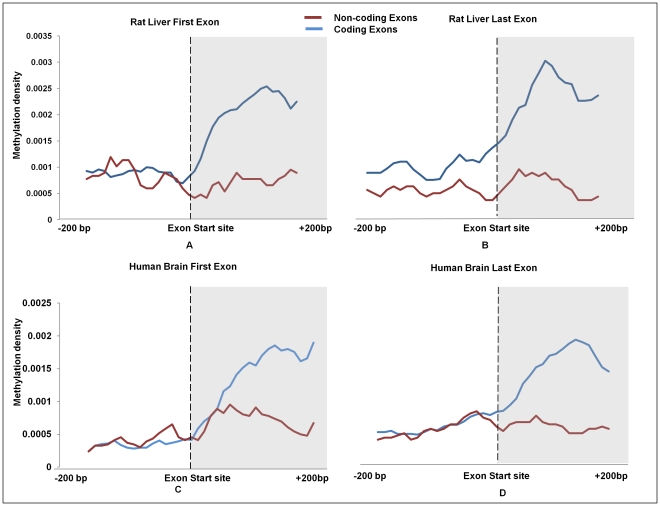
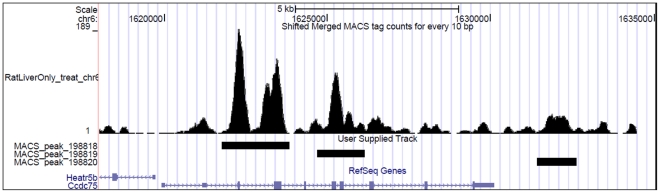
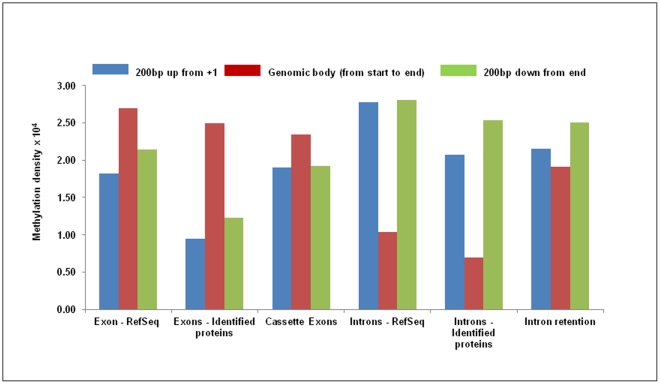
DNA methylation is crucial for gene regulation and maintenance of genomic stability. Rat has been a key model system in understanding mammalian systemic physiology, however detailed rat methylome remains uncharacterized till date. Here, we present the first high resolution methylome of rat liver generated using Methylated DNA immunoprecipitation and high throughput sequencing (MeDIP-Seq) approach. We observed that within the DNA/RNA repeat elements, simple repeats harbor the highest degree of methylation. Promoter hypomethylation and exon hypermethylation were common features in both RefSeq genes and expressed genes (as evaluated by proteomic approach). We also found that although CpG islands were generally hypomethylated, about 6% of them were methylated and a large proportion (37%) of methylated islands fell within the exons. Notably, we obeserved significant differences in methylation of terminal exons (UTRs); methylation being more pronounced in coding/partially coding exons compared to the non-coding exons. Further, events like alternate exon splicing (cassette exon) and intron retentions were marked by DNA methylation and these regions are retained in the final transcript. Thus, we suggest that DNA methylation could play a crucial role in marking coding regions thereby regulating alternative splicing. Apart from generating the first high resolution methylome map of rat liver tissue, the present study provides several critical insights into methylome organization and extends our understanding of interplay between epigenome, gene expression and genome stability.
Conflict of interest statement
Figures
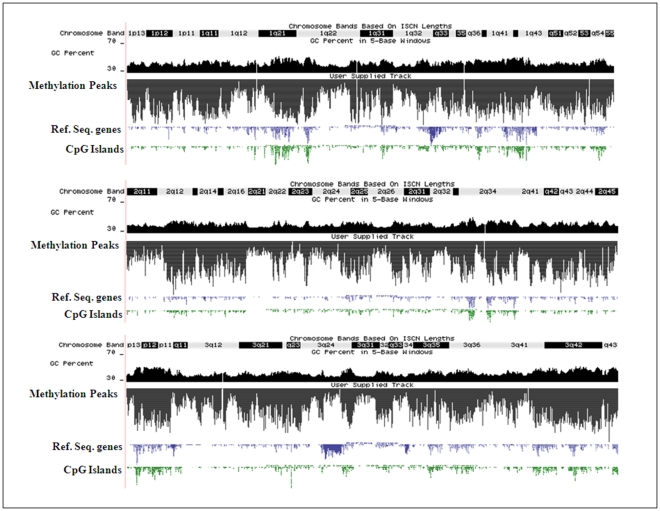
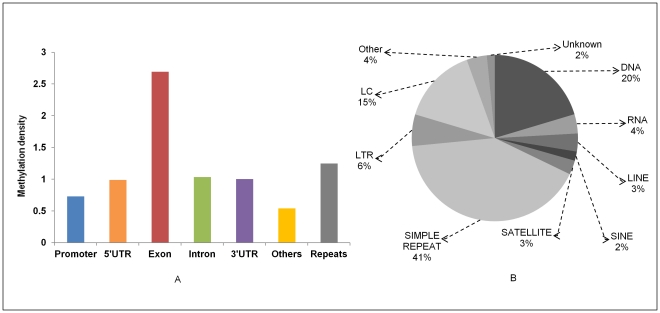
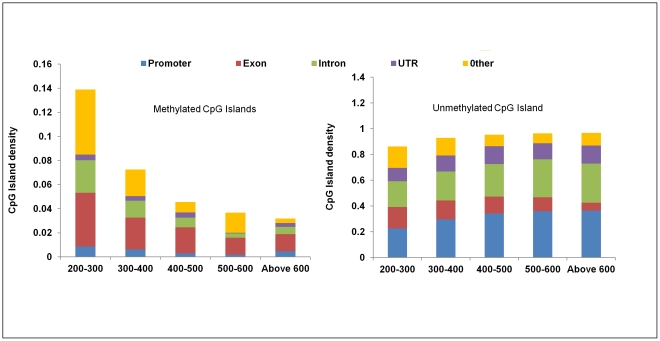
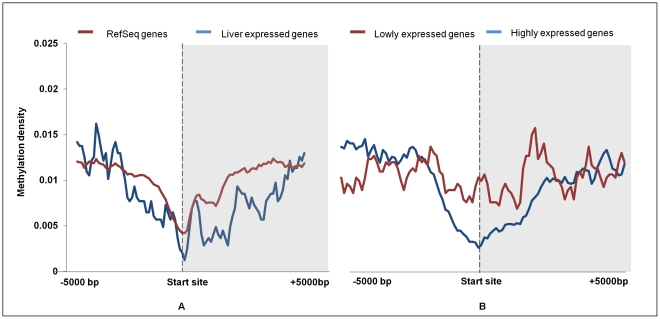
References
-
- Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–681. - PubMed
-
- Szyf M. The dynamic epigenome and its implications in toxicology. Toxicol Sci. 2007;100:7–23. - PubMed
-
- Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. - PubMed
-
- Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;465:721–727. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
