

The infinitely many genes model for the distributed genome of bacteria
- PMID: 22357598
- PMCID: PMC3342869
- DOI: 10.1093/gbe/evs016
The infinitely many genes model for the distributed genome of bacteria
Abstract
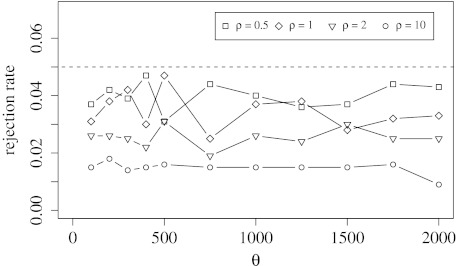
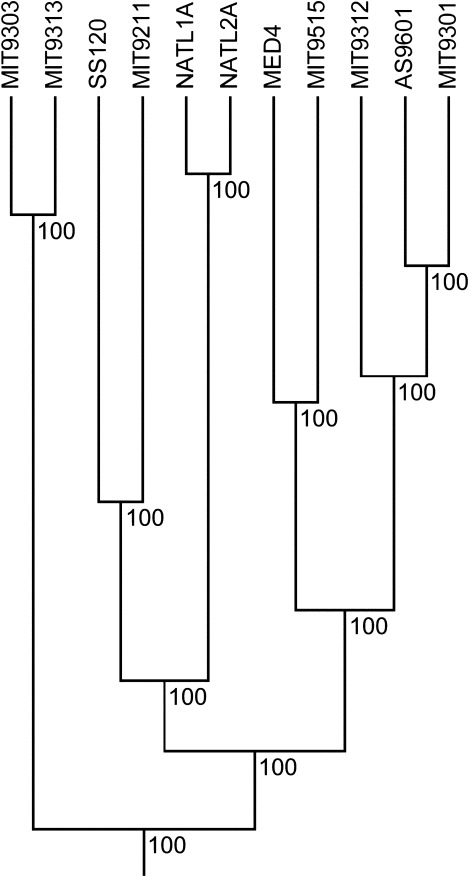
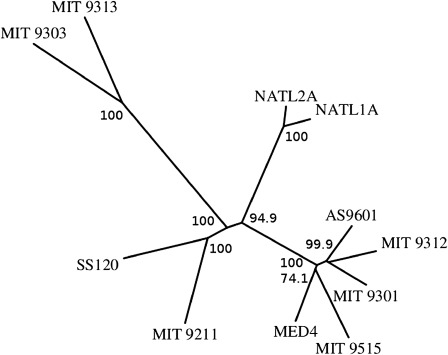
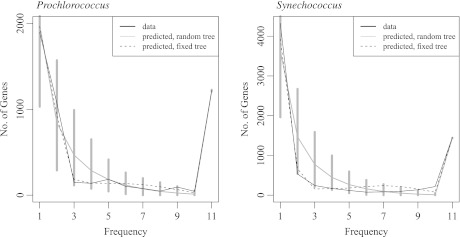
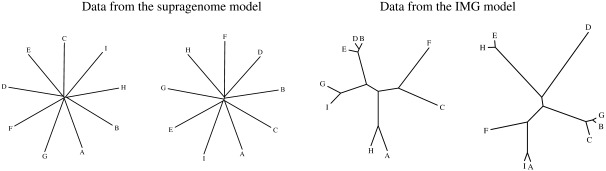
The distributed genome hypothesis states that the gene pool of a bacterial taxon is much more complex than that found in a single individual genome. However, the possible fitness advantage, why such genomic diversity is maintained, whether this variation is largely adaptive or neutral, and why these distinct individuals can coexist, remains poorly understood. Here, we present the infinitely many genes (IMG) model, which is a quantitative, evolutionary model for the distributed genome. It is based on a genealogy of individual genomes and the possibility of gene gain (from an unbounded reservoir of novel genes, e.g., by horizontal gene transfer from distant taxa) and gene loss, for example, by pseudogenization and deletion of genes, during reproduction. By implementing these mechanisms, the IMG model differs from existing concepts for the distributed genome, which cannot differentiate between neutral evolution and adaptation as drivers of the observed genomic diversity. Using the IMG model, we tested whether the distributed genome of 22 full genomes of picocyanobacteria (Prochlorococcus and Synechococcus) shows signs of adaptation or neutrality. We calculated the effective population size of Prochlorococcus at 1.01 × 10(11) and predicted 18 distinct clades for this population, only six of which have been isolated and cultured thus far. We predicted that the Prochlorococcus pangenome contains 57,792 genes and found that the evolution of the distributed genome of Prochlorococcus was possibly neutral, whereas that of Synechococcus and the combined sample shows a clear deviation from neutrality.
Figures
References
-
- Aldous D, Popovic L. A critical branching process model for biodiversity. Adv Appl Probab. 2005;37:1094–1115.
-
- Avrani S, Wurtzel O, Sharon I, Sorek R, Lindell D. Genomic island variability facilitates Prochlorococcus-virus coexistence. Nature. 2011;474:604–608. - PubMed
-
- Baumdicker F, Hess WR, Pfaffelhuber P. The diversity of a distributed genome in bacterial populations. Ann Appl Probab. 2010;20:1567–1606.
-
- Baumdicker F, Pfaffelhuber P. Evolution of bacterial genomes under horizontal gene transfer [Internet] 2011. Dublin (Ireland): ISI Congress. Available from: http://arxiv.org/abs/1105.5014, 1–8.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
