

DOGS: reaction-driven de novo design of bioactive compounds
- PMID: 22359493
- PMCID: PMC3280956
- DOI: 10.1371/journal.pcbi.1002380
DOGS: reaction-driven de novo design of bioactive compounds
Abstract
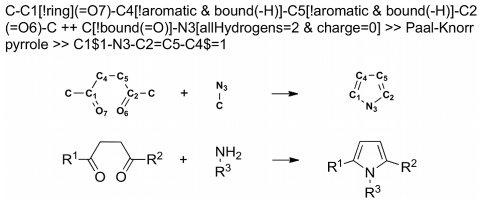
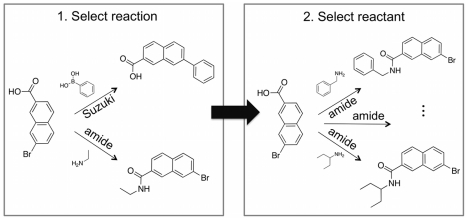
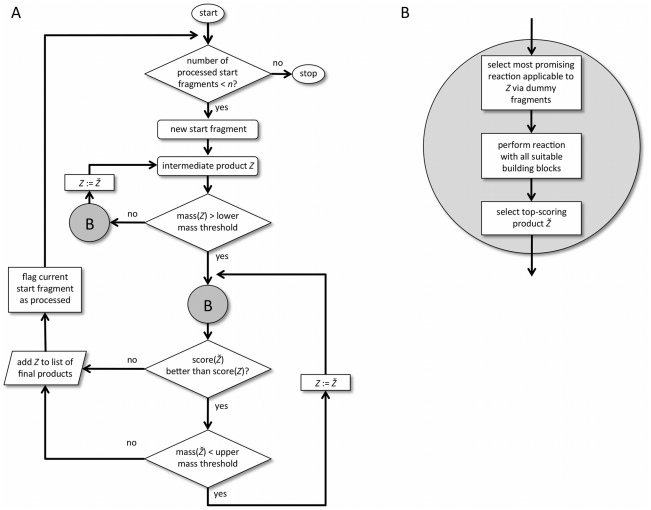
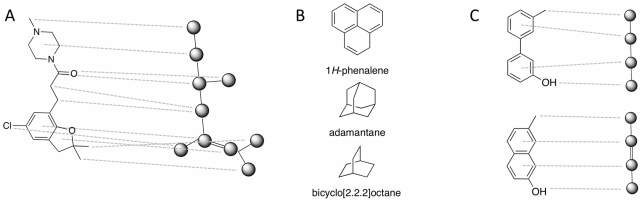
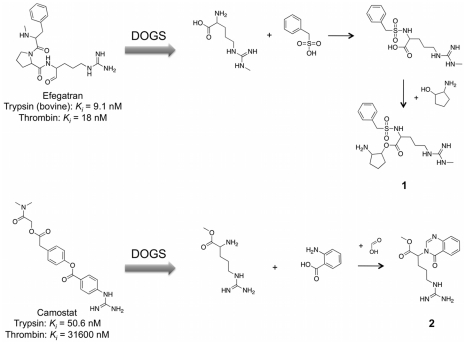
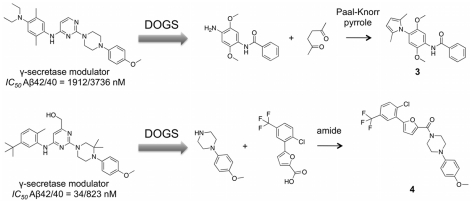
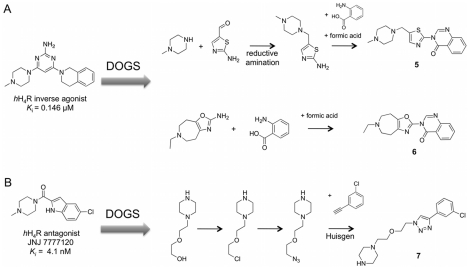
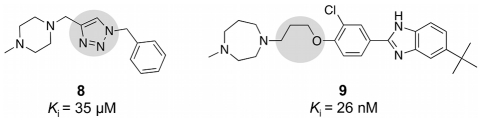
We present a computational method for the reaction-based de novo design of drug-like molecules. The software DOGS (Design of Genuine Structures) features a ligand-based strategy for automated 'in silico' assembly of potentially novel bioactive compounds. The quality of the designed compounds is assessed by a graph kernel method measuring their similarity to known bioactive reference ligands in terms of structural and pharmacophoric features. We implemented a deterministic compound construction procedure that explicitly considers compound synthesizability, based on a compilation of 25'144 readily available synthetic building blocks and 58 established reaction principles. This enables the software to suggest a synthesis route for each designed compound. Two prospective case studies are presented together with details on the algorithm and its implementation. De novo designed ligand candidates for the human histamine H₄ receptor and γ-secretase were synthesized as suggested by the software. The computational approach proved to be suitable for scaffold-hopping from known ligands to novel chemotypes, and for generating bioactive molecules with drug-like properties.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Discovery of novel histamine H4 and serotonin transporter ligands using the topological feature tree descriptor.J Chem Inf Model. 2012 Jan 23;52(1):233-42. doi: 10.1021/ci2004972. Epub 2011 Dec 28. J Chem Inf Model. 2012. PMID: 22168379
-
Homology model adjustment and ligand screening with a pseudoreceptor of the human histamine H4 receptor.ChemMedChem. 2009 May;4(5):820-7. doi: 10.1002/cmdc.200800443. ChemMedChem. 2009. PMID: 19343764
-
Scaffold-hopping potential of fragment-based de novo design: the chances and limits of variation.Comb Chem High Throughput Screen. 2009 May;12(4):383-96. doi: 10.2174/138620709788167971. Comb Chem High Throughput Screen. 2009. PMID: 19442066 Review.
-
Generate what you can make: achieving in-house synthesizability with readily available resources in de novo drug design.J Cheminform. 2025 Mar 28;17(1):41. doi: 10.1186/s13321-024-00910-4. J Cheminform. 2025. PMID: 40155970 Free PMC article.
-
Recent developments in de novo design and scaffold hopping.Curr Opin Drug Discov Devel. 2008 May;11(3):365-74. Curr Opin Drug Discov Devel. 2008. PMID: 18428090 Review.
Cited by
-
Ring system-based chemical graph generation for de novo molecular design.J Comput Aided Mol Des. 2016 May;30(5):425-46. doi: 10.1007/s10822-016-9916-1. Epub 2016 Jun 14. J Comput Aided Mol Des. 2016. PMID: 27299746
-
ReMODE: a deep learning-based web server for target-specific drug design.J Cheminform. 2022 Dec 12;14(1):84. doi: 10.1186/s13321-022-00665-w. J Cheminform. 2022. PMID: 36510307 Free PMC article.
-
QSAR without borders.Chem Soc Rev. 2020 Jun 7;49(11):3525-3564. doi: 10.1039/d0cs00098a. Epub 2020 May 1. Chem Soc Rev. 2020. PMID: 32356548 Free PMC article. Review.
-
Cheminformatics in Natural Product-based Drug Discovery.Mol Inform. 2020 Dec;39(12):e2000171. doi: 10.1002/minf.202000171. Epub 2020 Sep 6. Mol Inform. 2020. PMID: 32725781 Free PMC article. Review.
-
Identifying the macromolecular targets of de novo-designed chemical entities through self-organizing map consensus.Proc Natl Acad Sci U S A. 2014 Mar 18;111(11):4067-72. doi: 10.1073/pnas.1320001111. Epub 2014 Mar 3. Proc Natl Acad Sci U S A. 2014. PMID: 24591595 Free PMC article.
References
-
- Schneider G, Fechner U. Computer-based de novo design of druglike molecules. Nat Rev Drug Discov. 2005;4:649–663. - PubMed
-
- Hartenfeller M, Schneider G. De novo drug design. Methods Mol Biol. 2011;672:299–323. - PubMed
-
- Mauser H, Guba W. Recent developments in de novo design and scaffold hopping. Curr Opin Drug Discov Develop. 2008;11:365–374. - PubMed
-
- Kutchukian PS, Shakhnovich EI. De novo design: balancing novelty and confined chemical space. Expert Opin Drug Discov. 2010;5:789–812. - PubMed
-
- Boda K, Johnson AP. Molecular complexity analysis of de novo designed ligands. J Med Chem. 2006;49:5869–5879. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
