

Exon-specific QTLs skew the inferred distribution of expression QTLs detected using gene expression array data
- PMID: 22359548
- PMCID: PMC3281037
- DOI: 10.1371/journal.pone.0030629
Exon-specific QTLs skew the inferred distribution of expression QTLs detected using gene expression array data
Abstract
Mapping of expression quantitative trait loci (eQTLs) is an important technique for studying how genetic variation affects gene regulation in natural populations. In a previous study using Illumina expression data from human lymphoblastoid cell lines, we reported that cis-eQTLs are especially enriched around transcription start sites (TSSs) and immediately upstream of transcription end sites (TESs). In this paper, we revisit the distribution of eQTLs using additional data from Affymetrix exon arrays and from RNA sequencing. We confirm that most eQTLs lie close to the target genes; that transcribed regions are generally enriched for eQTLs; that eQTLs are more abundant in exons than introns; and that the peak density of eQTLs occurs at the TSS. However, we find that the intriguing TES peak is greatly reduced or absent in the Affymetrix and RNA-seq data. Instead our data suggest that the TES peak observed in the Illumina data is mainly due to exon-specific QTLs that affect 3' untranslated regions, where most of the Illumina probes are positioned. Nonetheless, we do observe an overall enrichment of eQTLs in exons versus introns in all three data sets, consistent with an important role for exonic sequences in gene regulation.
Conflict of interest statement
Figures
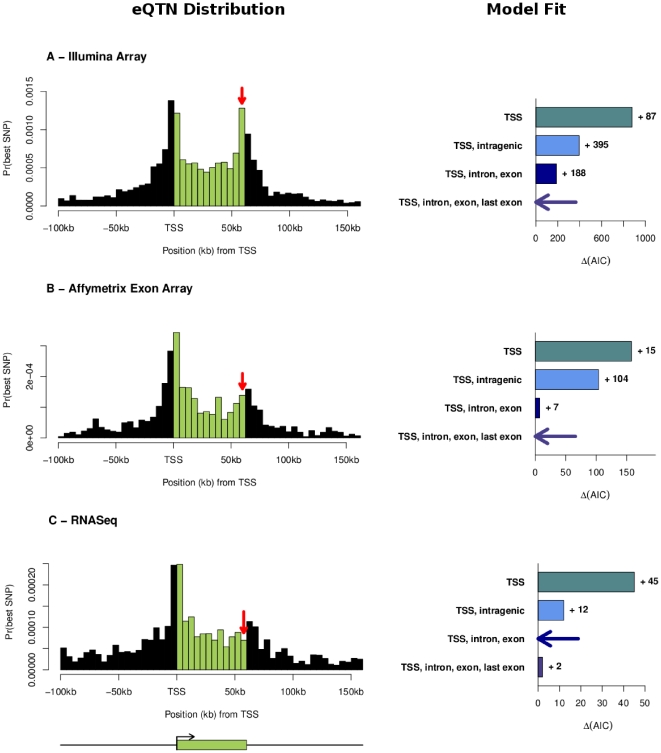
 “(AIC”) indicate better model fit, and the best model for each data set is indicated with a horizontal arrow. The labels for the four models indicate the different parameters included in each model: “TSS” refers to our basic distance model measured as distance from TSS; “intragenic” means that we use a single additional parameter for all SNPs within the transcript; “exon, intron” indicates that we use separate parameters for exonic and intronic SNPs respectively, and “last exon” indicates that we add an additional parameter for SNPs in the final exon.
“(AIC”) indicate better model fit, and the best model for each data set is indicated with a horizontal arrow. The labels for the four models indicate the different parameters included in each model: “TSS” refers to our basic distance model measured as distance from TSS; “intragenic” means that we use a single additional parameter for all SNPs within the transcript; “exon, intron” indicates that we use separate parameters for exonic and intronic SNPs respectively, and “last exon” indicates that we add an additional parameter for SNPs in the final exon.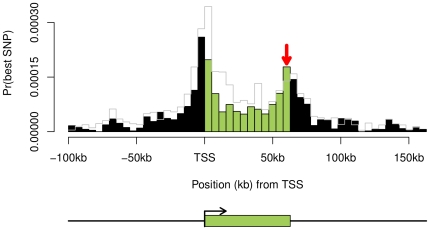
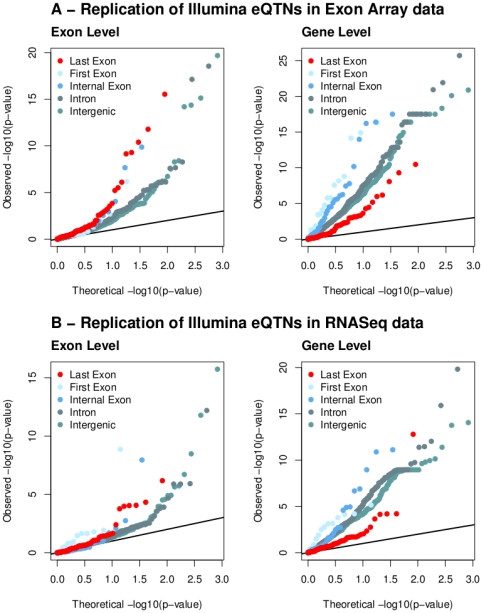
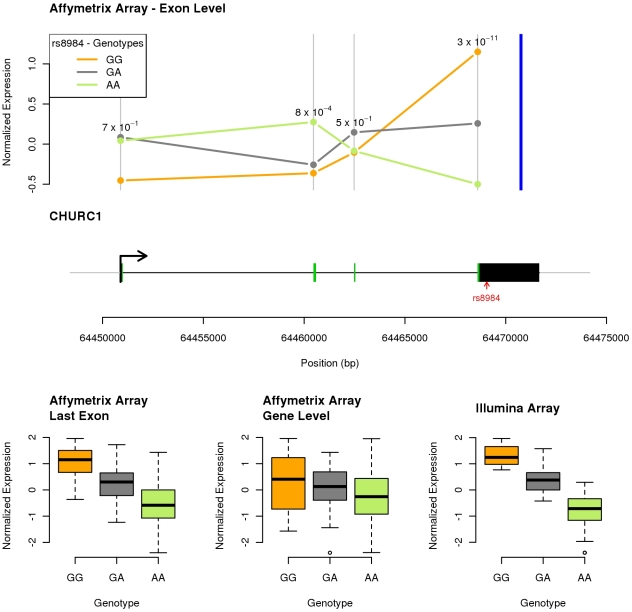
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
