

Deciphering the preference and predicting the viability of circular permutations in proteins
- PMID: 22359629
- PMCID: PMC3281007
- DOI: 10.1371/journal.pone.0031791
Deciphering the preference and predicting the viability of circular permutations in proteins
Abstract
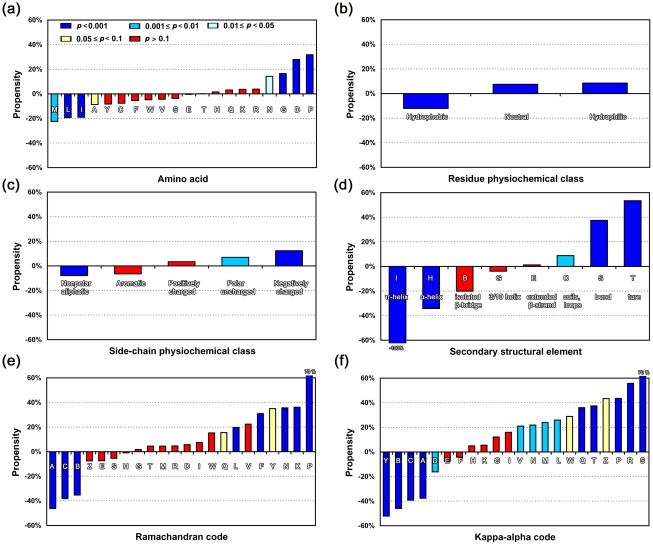
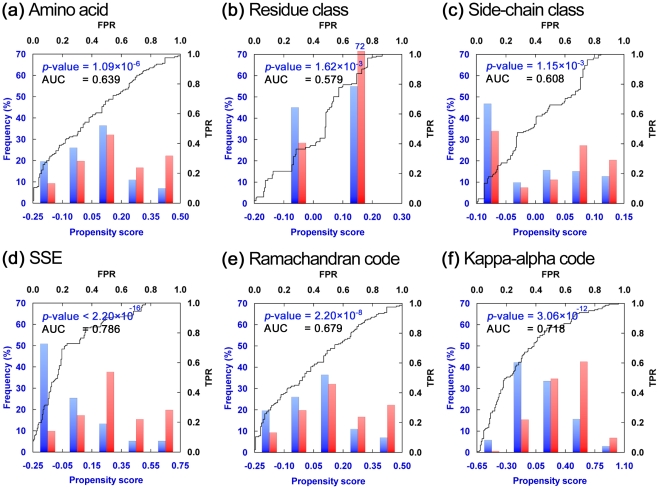
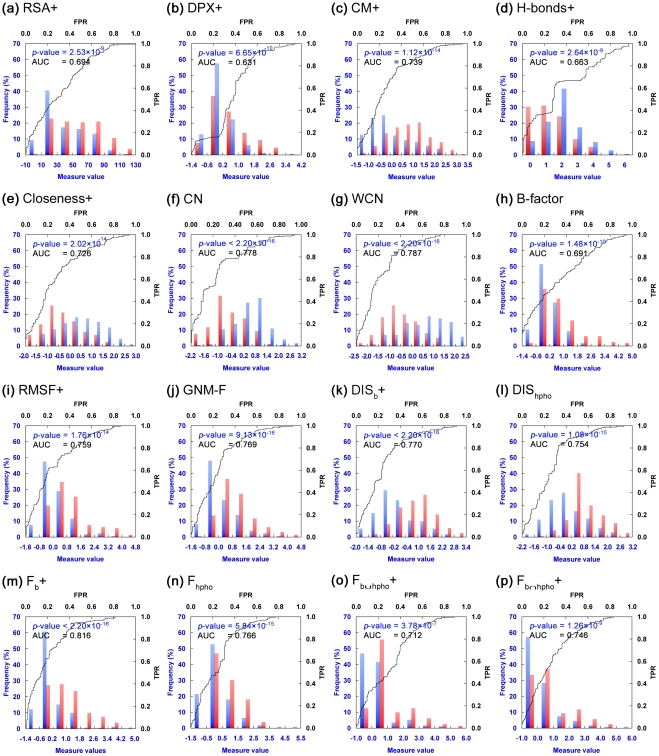
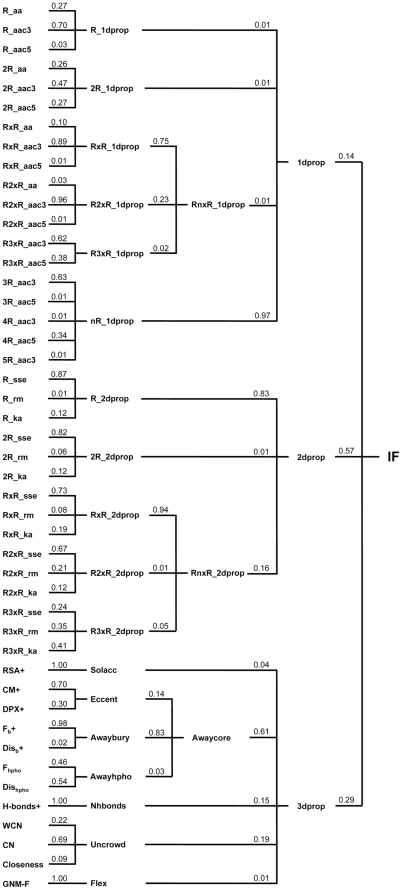
Circular permutation (CP) refers to situations in which the termini of a protein are relocated to other positions in the structure. CP occurs naturally and has been artificially created to study protein function, stability and folding. Recently CP is increasingly applied to engineer enzyme structure and function, and to create bifunctional fusion proteins unachievable by tandem fusion. CP is a complicated and expensive technique. An intrinsic difficulty in its application lies in the fact that not every position in a protein is amenable for creating a viable permutant. To examine the preferences of CP and develop CP viability prediction methods, we carried out comprehensive analyses of the sequence, structural, and dynamical properties of known CP sites using a variety of statistics and simulation methods, such as the bootstrap aggregating, permutation test and molecular dynamics simulations. CP particularly favors Gly, Pro, Asp and Asn. Positions preferred by CP lie within coils, loops, turns, and at residues that are exposed to solvent, weakly hydrogen-bonded, environmentally unpacked, or flexible. Disfavored positions include Cys, bulky hydrophobic residues, and residues located within helices or near the protein's core. These results fostered the development of an effective viable CP site prediction system, which combined four machine learning methods, e.g., artificial neural networks, the support vector machine, a random forest, and a hierarchical feature integration procedure developed in this work. As assessed by using the hydrofolate reductase dataset as the independent evaluation dataset, this prediction system achieved an AUC of 0.9. Large-scale predictions have been performed for nine thousand representative protein structures; several new potential applications of CP were thus identified. Many unreported preferences of CP are revealed in this study. The developed system is the best CP viability prediction method currently available. This work will facilitate the application of CP in research and biotechnology.
Conflict of interest statement
Figures
References
-
- Carrington DM, Auffret A, Hanke DE. Polypeptide ligation occurs during post-translational modification of concanavalin A. Nature. 1985;313:64–67. - PubMed
-
- Ponting CP, Russell RB. Swaposins: circular permutations within genes encoding saposin homologues. Trends Biochem Sci. 1995;20:179–180. - PubMed
-
- Lindqvist Y, Schneider G. Circular permutations of natural protein sequences: structural evidence. Curr Opin Struct Biol. 1997;7:422–427. - PubMed
-
- Uliel S, Fliess A, Unger R. Naturally occurring circular permutations in proteins. Protein Eng. 2001;14:533–542. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
