

Human anti-prion antibodies block prion peptide fibril formation and neurotoxicity
- PMID: 22362783
- PMCID: PMC3339962
- DOI: 10.1074/jbc.M111.255836
Human anti-prion antibodies block prion peptide fibril formation and neurotoxicity
Abstract
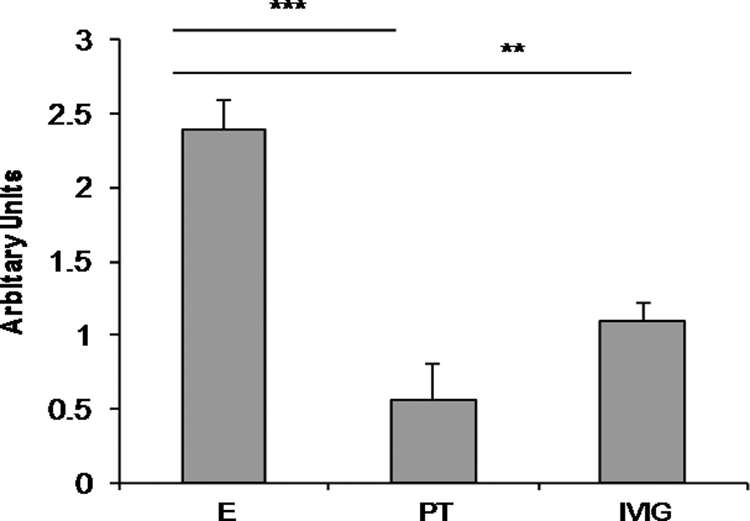
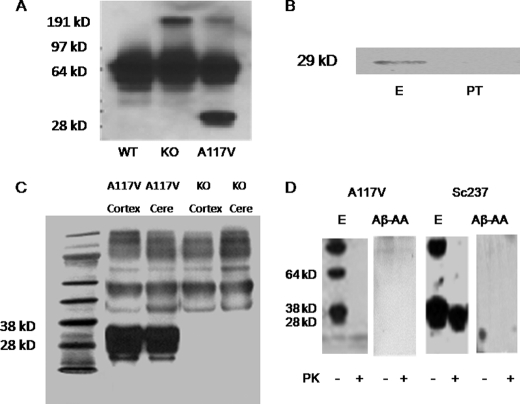
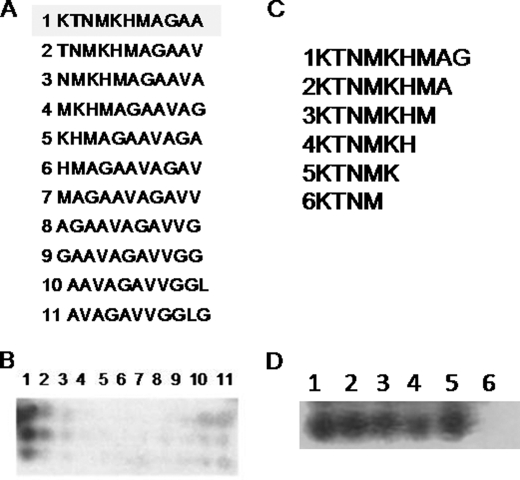
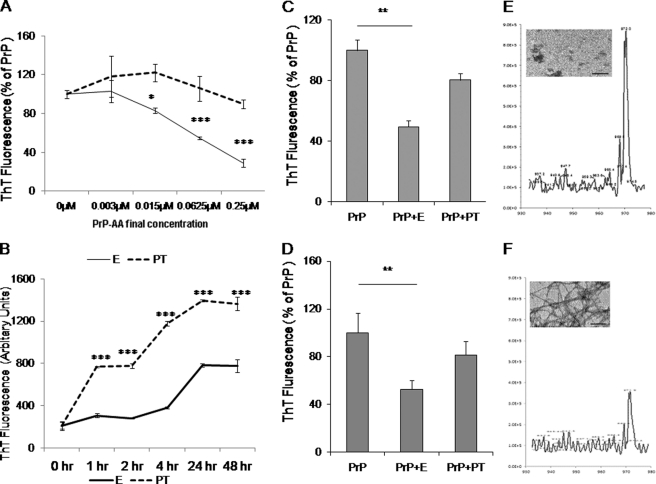
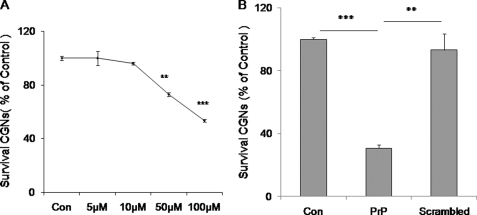
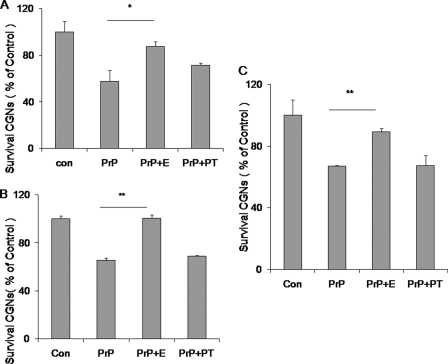
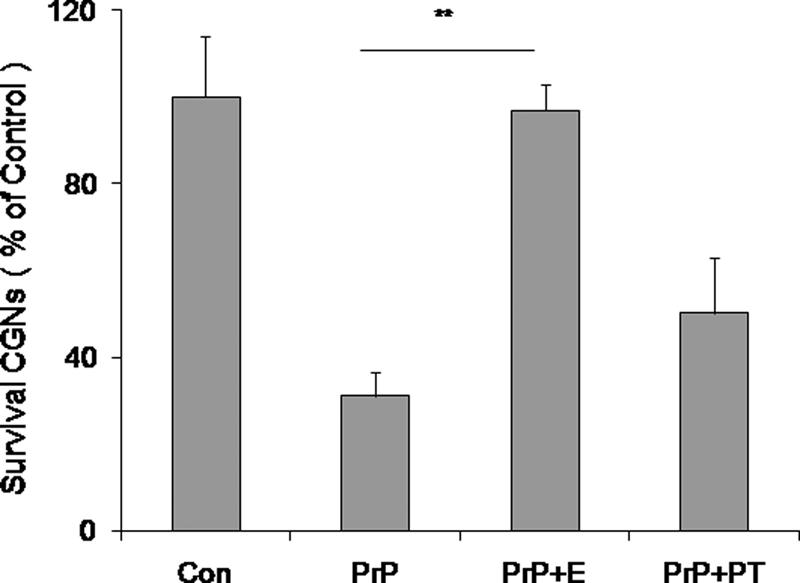
Prion diseases are a group of rare, fatal neurodegenerative disorders associated with a conformational transformation of the cellular prion protein (PrP(C)) into a self-replicating and proteinase K-resistant conformer, termed scrapie PrP (PrP(Sc)). Aggregates of PrP(Sc) deposited around neurons lead to neuropathological alterations. Currently, there is no effective treatment for these fatal illnesses; thus, the development of an effective therapy is a priority. PrP peptide-based ELISA assay methods were developed for detection and immunoaffinity chromatography capture was developed for purification of naturally occurring PrP peptide autoantibodies present in human CSF, individual donor serum, and commercial preparations of pooled intravenous immunoglobulin (IVIg). The ratio of anti-PrP autoantibodies (PrP-AA) to total IgG was ∼1:1200. The binding epitope of purified PrP-AA was mapped to an N-terminal region comprising the PrP amino acid sequence KTNMK. Purified PrP-AA potently blocked fibril formation by a toxic 21-amino acid fragment of the PrP peptide containing the amino acid alanine to valine substitution corresponding to position 117 of the full-length peptide (A117V). Furthermore, PrP-AA attenuated the neurotoxicity of PrP(A117V) and wild-type peptides in rat cerebellar granule neuron (CGN) cultures. In contrast, IgG preparations depleted of PrP-AA had little effect on PrP fibril formation or PrP neurotoxicity. The specificity of PrP-AA was demonstrated by immunoprecipitating PrP protein in brain tissues of transgenic mice expressing the human PrP(A117V) epitope and Sc237 hamster. Based on these intriguing findings, it is suggested that human PrP-AA may be useful for interfering with the pathogenic effects of pathogenic prion proteins and, thereby has the potential to be an effective means for preventing or attenuating human prion disease progression.
Figures
Similar articles
-
Vaccination with prion peptide-displaying papillomavirus-like particles induces autoantibodies to normal prion protein that interfere with pathologic prion protein production in infected cells.FEBS J. 2007 Apr;274(7):1747-58. doi: 10.1111/j.1742-4658.2007.05721.x. Epub 2007 Feb 20. FEBS J. 2007. PMID: 17313482 Free PMC article.
-
Motif-grafted antibodies containing the replicative interface of cellular PrP are specific for PrPSc.Proc Natl Acad Sci U S A. 2004 Jul 13;101(28):10404-9. doi: 10.1073/pnas.0403522101. Epub 2004 Jul 6. Proc Natl Acad Sci U S A. 2004. PMID: 15240877 Free PMC article.
-
Clearance and prevention of prion infection in cell culture by anti-PrP antibodies.Eur J Neurosci. 2006 May;23(10):2635-47. doi: 10.1111/j.1460-9568.2006.04805.x. Eur J Neurosci. 2006. PMID: 16817866 Free PMC article.
-
Aptamers against prion proteins and prions.Cell Mol Life Sci. 2009 Aug;66(15):2445-55. doi: 10.1007/s00018-009-0031-5. Epub 2009 Apr 25. Cell Mol Life Sci. 2009. PMID: 19396399 Free PMC article. Review.
-
Neurometals in the Pathogenesis of Prion Diseases.Int J Mol Sci. 2021 Jan 28;22(3):1267. doi: 10.3390/ijms22031267. Int J Mol Sci. 2021. PMID: 33525334 Free PMC article. Review.
Cited by
-
Single-chain fragment variable passive immunotherapies for neurodegenerative diseases.Int J Mol Sci. 2013 Sep 17;14(9):19109-27. doi: 10.3390/ijms140919109. Int J Mol Sci. 2013. PMID: 24048248 Free PMC article. Review.
-
Immunotherapy in prion disease.Nat Rev Neurol. 2013 Feb;9(2):98-105. doi: 10.1038/nrneurol.2012.258. Epub 2012 Dec 18. Nat Rev Neurol. 2013. PMID: 23247613 Review.
-
Brain iron homeostasis: from molecular mechanisms to clinical significance and therapeutic opportunities.Antioxid Redox Signal. 2014 Mar 10;20(8):1324-63. doi: 10.1089/ars.2012.4931. Epub 2013 Aug 15. Antioxid Redox Signal. 2014. PMID: 23815406 Free PMC article. Review.
-
IVIG Delays Onset in a Mouse Model of Gerstmann-Sträussler-Scheinker Disease.Mol Neurobiol. 2019 Apr;56(4):2353-2361. doi: 10.1007/s12035-018-1228-0. Epub 2018 Jul 19. Mol Neurobiol. 2019. PMID: 30027340 Free PMC article.
-
Immunization of cervidized transgenic mice with multimeric deer prion protein induces self-antibodies that antagonize chronic wasting disease infectivity in vitro.Sci Rep. 2017 Sep 5;7(1):10538. doi: 10.1038/s41598-017-11235-8. Sci Rep. 2017. PMID: 28874781 Free PMC article.
References
-
- Prusiner S. B. (1991) Molecular biology of prion diseases. Science 252, 1515–1522 - PubMed
-
- Bugiani O., Giaccone G., Piccardo P., Morbin M., Tagliavini F., Ghetti B. (2000) Neuropathology of Gerstmann-Sträussler-Scheinker disease. Microsc. Res. Tech. 50, 10–15 - PubMed
-
- Ghetti B., Piccardo P., Frangione B., Bugiani O., Giaccone G., Young K., Prelli F., Farlow M. R., Dlouhy S. R., Tagliavini F. (1996) Prion protein amyloidosis. Brain Pathol. 6, 127–145 - PubMed
-
- Kitamoto T., Tateishi J., Tashima T., Takeshita I., Barry R. A., DeArmond S. J., Prusiner S. B. (1986) Amyloid plaques in Creutzfeldt-Jakob disease stain with prion protein antibodies. Ann. Neurol. 20, 204–208 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
