

Tracing lifestyle adaptation in prokaryotic genomes
- PMID: 22363326
- PMCID: PMC3282942
- DOI: 10.3389/fmicb.2012.00048
Tracing lifestyle adaptation in prokaryotic genomes
Abstract
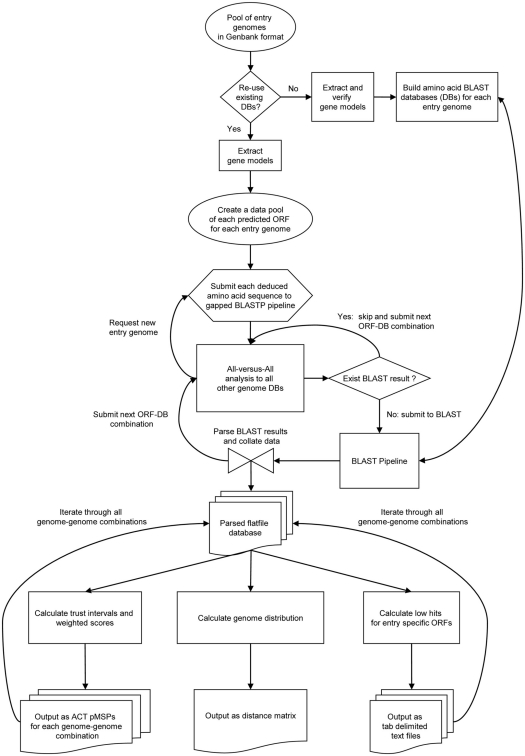
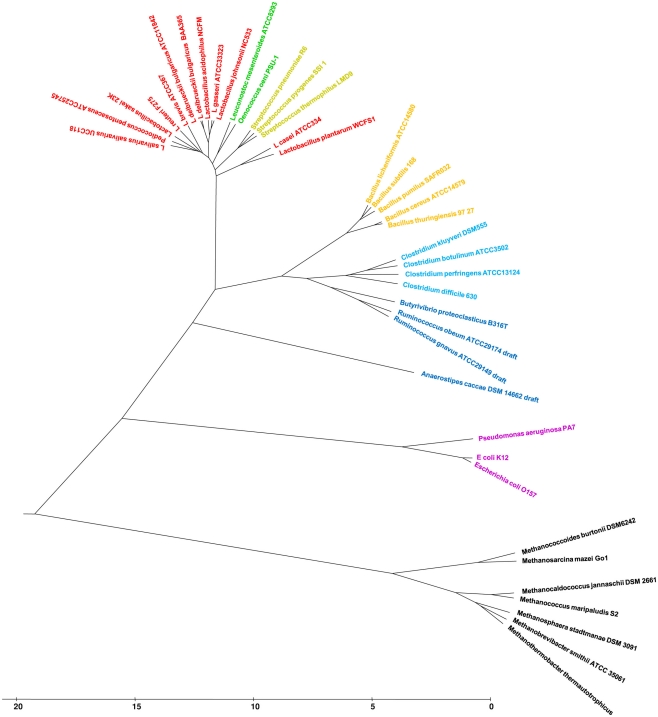
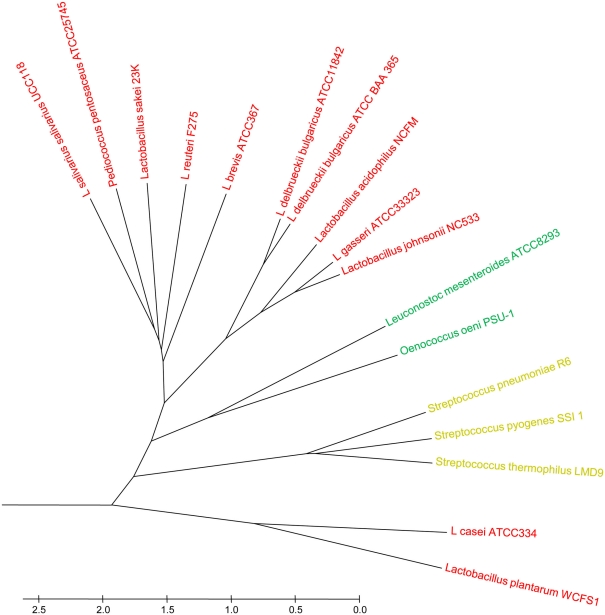
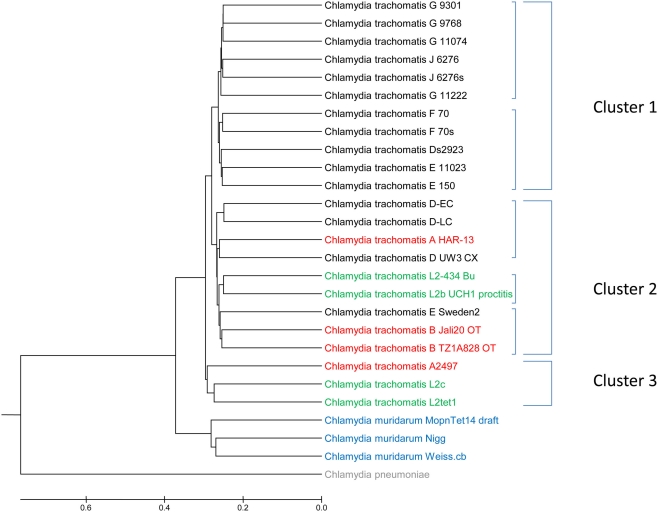
Lifestyle adaptation of microbes due to changes in their ecological niches or acquisition of new environments is a major driving force for genetic changes in their respective genomes. Moving into more specialized niches often results in the acquisition of new gene sets via horizontal gene transfer to utilize previously unavailable metabolites, while genetic ballast is shed by gene loss and/or gene inactivation. In some cases, larger genome rearrangements can be observed, such as the incorporation of whole genetic islands, providing a range of new phenotypic capabilities. Until recently these changes could not be comprehensively followed and identified due to the lack of complete microbial genome sequences. The advent of high-throughput DNA sequencing has dramatically changed the scientific landscape and today microbial genomes have become increasingly abundant. Currently, more than 2,900 genomes are published and more than 11,000 genome projects are listed in the Genomes Online Database. Although this wealth of information provides many new opportunities to assess microbial functionality, it also creates a new array of challenges when a comparison between multiple microbial genomes is required. Here, functional genome distribution (FGD) is introduced, analyzing the diversity between microbes based on their predicted ORFeome. FGD is therefore a comparative genomics approach, emphasizing the assessments of gene complements. To further facilitate the comparison between two or more genomes, degrees of amino-acid similarities between ORFeomes can be visualized in the Artemis comparison tool, graphically depicting small and large scale genome rearrangements, insertion and deletion events, and levels of similarity between individual open reading frames. FGD provides a new tool for comparative microbial genomics and the interpretation of differences in the genetic makeup of bacteria.
Keywords: functional genomics; genome comparison; genome plasticity; horizontal gene transfer; lifestyle adaption.
Figures

References
-
- Altermann E., Russell W. M., Azcarate-Peril M. A., Barrangou R., Buck B. L., Mcauliffe O., Souther N., Dobson A., Duong T., Callanan M., Lick S., Hamrick A., Cano R., Klaenhammer T. R. (2005). Complete genome sequence of the probiotic lactic acid bacterium Lactobacillus acidophilus NCFM. Proc. Natl. Acad. Sci. U.S.A. 102, 3906–3912 10.1073/pnas.0409188102 - DOI - PMC - PubMed
-
- Azcarate-Peril M. A., Altermann E., Goh Y. J., Tallon R., Sanozky-Dawes R. B., Pfeiler E. A., O’flaherty S., Buck B. L., Dobson A., Duong T., Miller M. J., Barrangou R., Klaenhammer T. R. (2008). Analysis of the genome sequence of Lactobacillus gasseri ATCC 33323 reveals the molecular basis of an autochthonous intestinal organism. Appl. Environ. Microbiol. 74, 4610–4625 10.1128/AEM.00054-08 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases