

Yersinia pestis lineages in Mongolia
- PMID: 22363455
- PMCID: PMC3281858
- DOI: 10.1371/journal.pone.0030624
Yersinia pestis lineages in Mongolia
Abstract
Background: Whole genome sequencing allowed the development of a number of high resolution sequence based typing tools for Yersinia (Y.) pestis. The application of these methods on isolates from most known foci worldwide and in particular from China and the Former Soviet Union has dramatically improved our understanding of the population structure of this species. In the current view, Y. pestis including the non or moderate human pathogen Y. pestis subspecies microtus emerged from Yersinia pseudotuberculosis about 2,600 to 28,600 years ago in central Asia. The majority of central Asia natural foci have been investigated. However these investigations included only few strains from Mongolia.
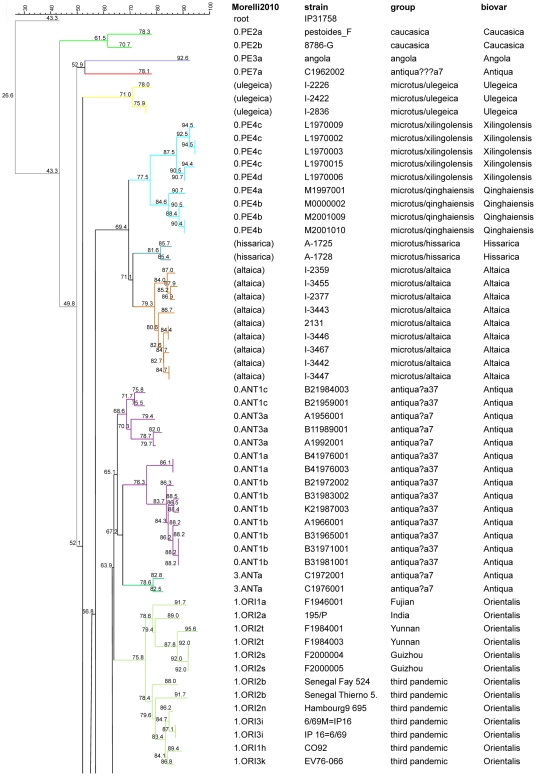
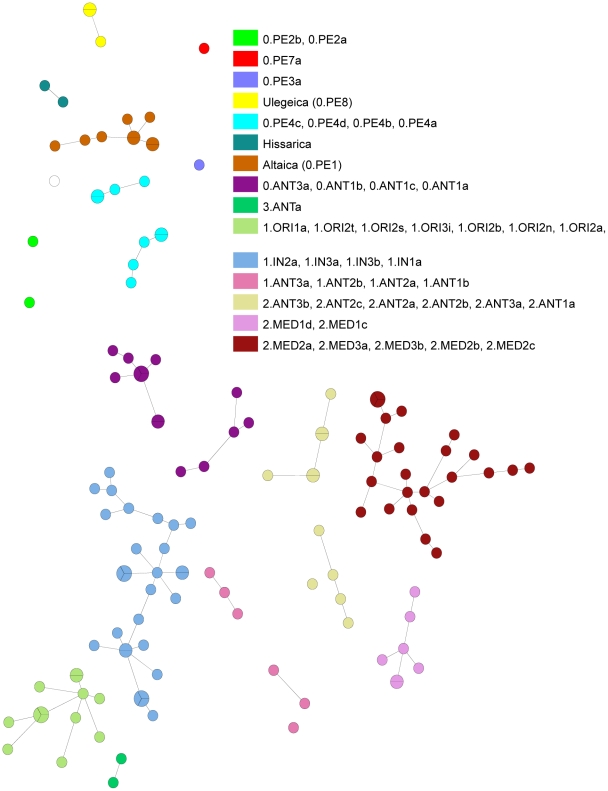
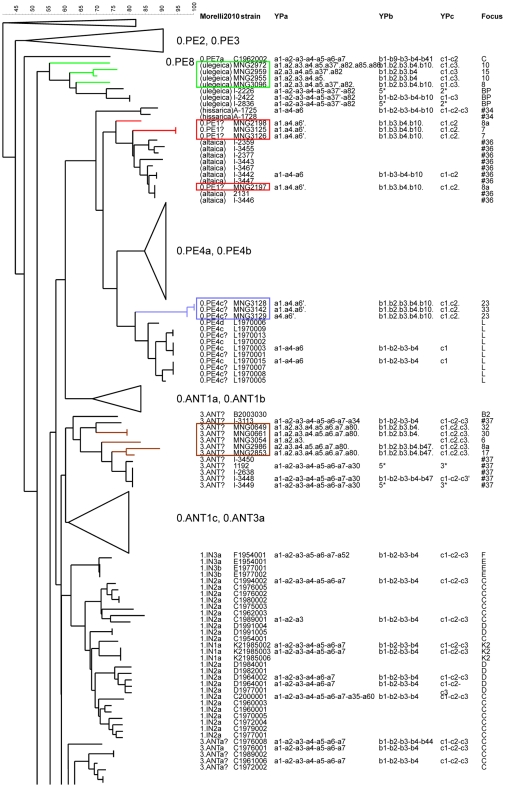
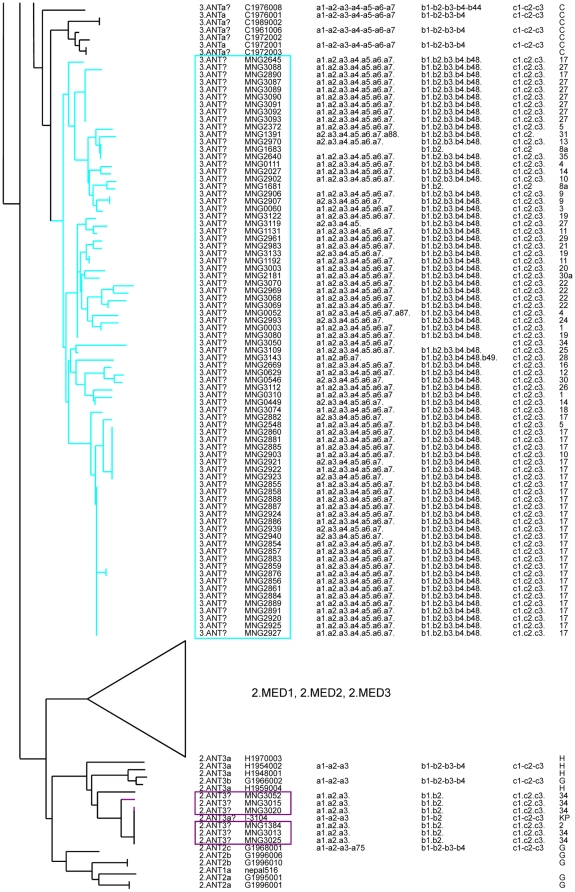
Methodology/principal findings: Clustered Regularly Interspaced Short Prokaryotic Repeats (CRISPR) analysis and Multiple-locus variable number of tandem repeats (VNTR) analysis (MLVA) with 25 loci was performed on 100 Y. pestis strains, isolated from 37 sampling areas in Mongolia. The resulting data were compared with previously published data from more than 500 plague strains, 130 of which had also been previously genotyped by single nucleotide polymorphism (SNP) analysis. The comparison revealed six main clusters including the three microtus biovars Ulegeica, Altaica, and Xilingolensis. The largest cluster comprises 78 isolates, with unique and new genotypes seen so far in Mongolia only. Typing of selected isolates by key SNPs was used to robustly assign the corresponding clusters to previously defined SNP branches.
Conclusions/significance: We show that Mongolia hosts the most recent microtus clade (Ulegeica). Interestingly no representatives of the ancestral Y. pestis subspecies pestis nodes previously identified in North-western China were identified in this study. This observation suggests that the subsequent evolution steps within Y. pestis pestis did not occur in Mongolia. Rather, Mongolia was most likely re-colonized by more recent clades coming back from China contemporary of the black death pandemic, or more recently in the past 600 years.
Conflict of interest statement
Figures



Similar articles
-
Genotyping and phylogenetic analysis of Yersinia pestis by MLVA: insights into the worldwide expansion of Central Asia plague foci.PLoS One. 2009 Jun 22;4(6):e6000. doi: 10.1371/journal.pone.0006000. PLoS One. 2009. PMID: 19543392 Free PMC article.
-
[Standard algorithm of molecular typing of Yersinia pestis strains].Zh Mikrobiol Epidemiol Immunobiol. 2012 May-Jun;(3):25-35. Zh Mikrobiol Epidemiol Immunobiol. 2012. PMID: 22830271 Russian.
-
MLVA distribution characteristics of Yersinia pestis in China and the correlation analysis.BMC Microbiol. 2009 Sep 23;9:205. doi: 10.1186/1471-2180-9-205. BMC Microbiol. 2009. PMID: 19775435 Free PMC article.
-
Review of genotyping methods for Yersinia pestis in Madagascar.PLoS Negl Trop Dis. 2024 Jun 27;18(6):e0012252. doi: 10.1371/journal.pntd.0012252. eCollection 2024 Jun. PLoS Negl Trop Dis. 2024. PMID: 38935608 Free PMC article. Review.
-
Taxonomy of Yersinia pestis.Adv Exp Med Biol. 2016;918:35-78. doi: 10.1007/978-94-024-0890-4_3. Adv Exp Med Biol. 2016. PMID: 27722860 Review.
Cited by
-
Dynamics of CRISPR loci in microevolutionary process of Yersinia pestis strains.PLoS One. 2014 Sep 29;9(9):e108353. doi: 10.1371/journal.pone.0108353. eCollection 2014. PLoS One. 2014. PMID: 25265542 Free PMC article.
-
Biological activity evaluations of chemical constituents derived from Mongolian medicinal forage plants and their applications in combating infectious diseases and addressing health problems in humans and livestock.J Nat Med. 2021 Sep;75(4):729-740. doi: 10.1007/s11418-021-01529-7. Epub 2021 May 21. J Nat Med. 2021. PMID: 34018093 Free PMC article. Review.
-
Ten years of surveillance of the Yulong plague focus in China and the molecular typing and source tracing of the isolates.PLoS Negl Trop Dis. 2018 Mar 30;12(3):e0006352. doi: 10.1371/journal.pntd.0006352. eCollection 2018 Mar. PLoS Negl Trop Dis. 2018. PMID: 29601573 Free PMC article.
-
Yersinia pestis: the Natural History of Plague.Clin Microbiol Rev. 2020 Dec 9;34(1):e00044-19. doi: 10.1128/CMR.00044-19. Print 2020 Dec 16. Clin Microbiol Rev. 2020. PMID: 33298527 Free PMC article. Review.
-
Yersinia pestis DNA from skeletal remains from the 6(th) century AD reveals insights into Justinianic Plague.PLoS Pathog. 2013;9(5):e1003349. doi: 10.1371/journal.ppat.1003349. Epub 2013 May 2. PLoS Pathog. 2013. PMID: 23658525 Free PMC article.
References
-
- World Health Organization. Weekly epidemiological record. 2003;78:129–136.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
