

Anti-inflammatory role of the cAMP effectors Epac and PKA: implications in chronic obstructive pulmonary disease
- PMID: 22363678
- PMCID: PMC3283666
- DOI: 10.1371/journal.pone.0031574
Anti-inflammatory role of the cAMP effectors Epac and PKA: implications in chronic obstructive pulmonary disease
Abstract
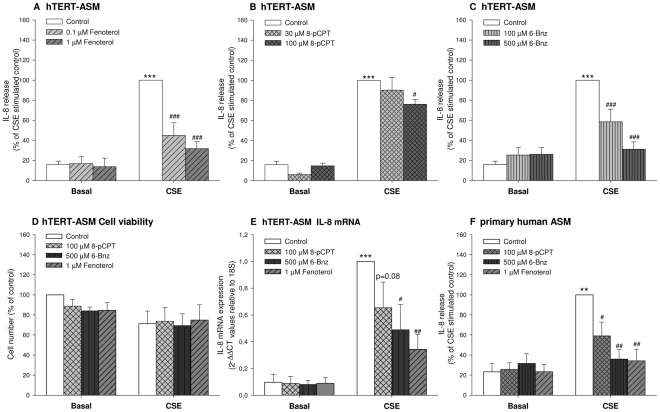
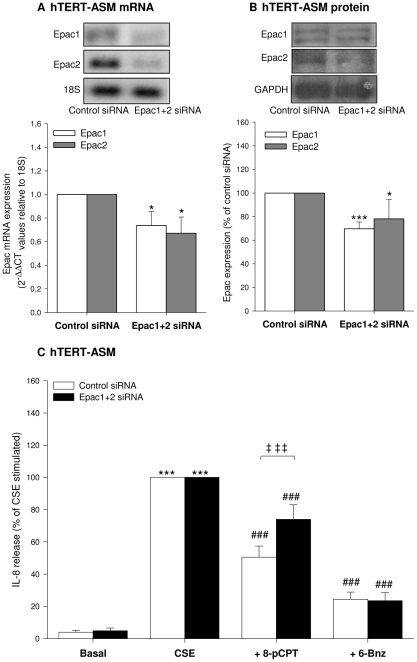
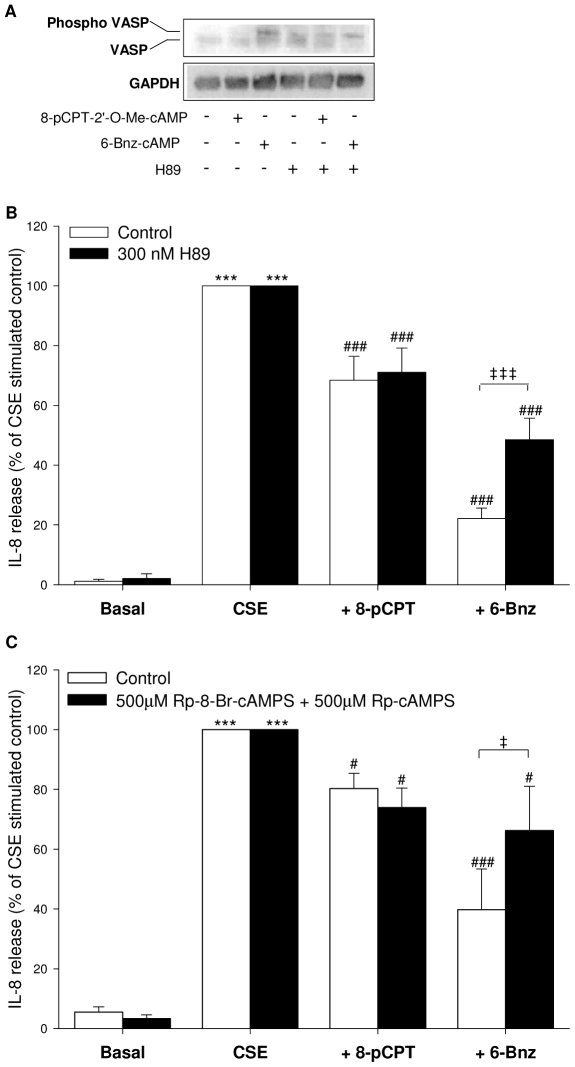
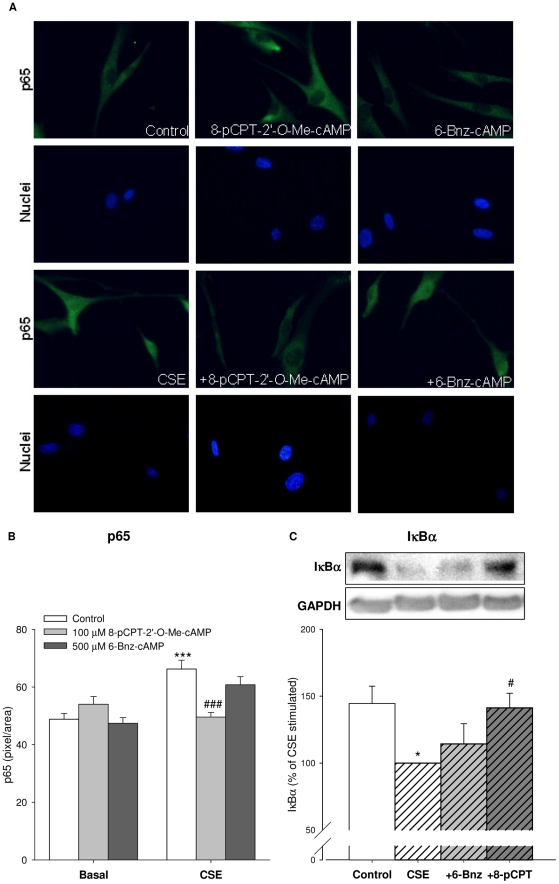
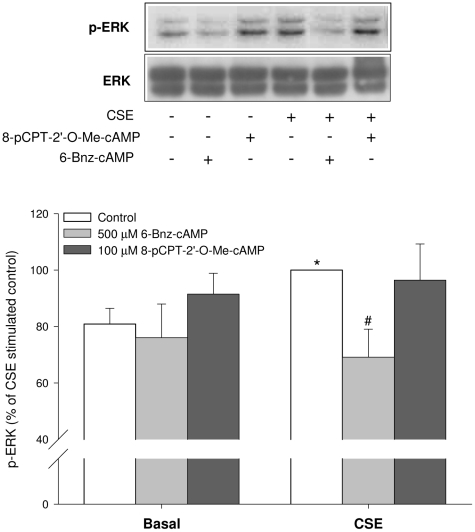
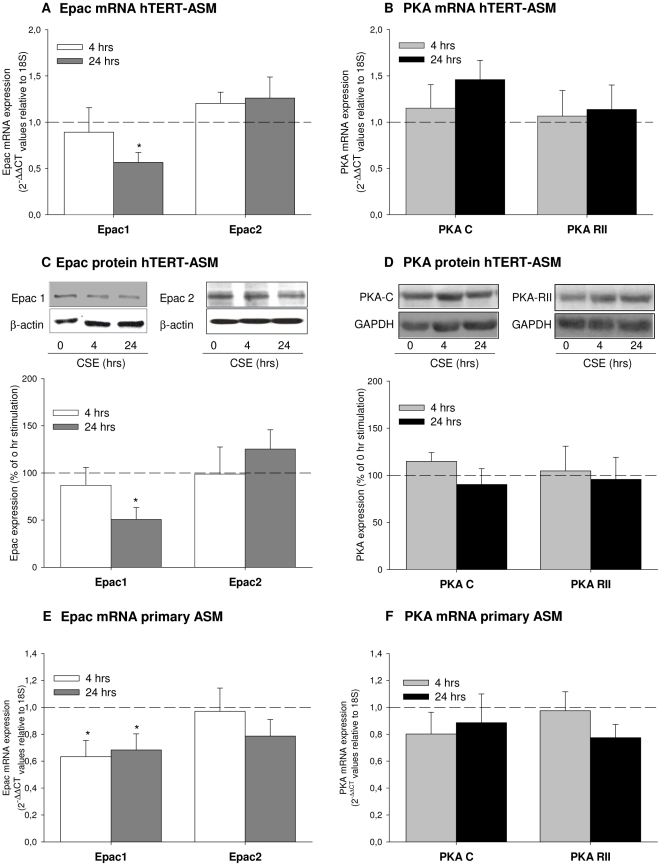
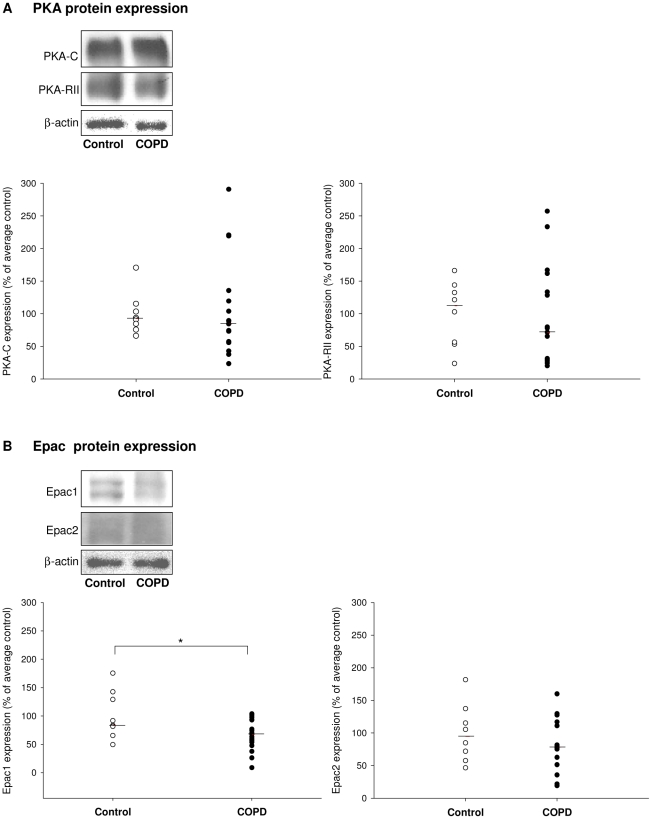
Cigarette smoke-induced release of pro-inflammatory cytokines including interleukin-8 (IL-8) from inflammatory as well as structural cells in the airways, including airway smooth muscle (ASM) cells, may contribute to the development of chronic obstructive pulmonary disease (COPD). Despite the wide use of pharmacological treatment aimed at increasing intracellular levels of the endogenous suppressor cyclic AMP (cAMP), little is known about its exact mechanism of action. We report here that next to the β(2)-agonist fenoterol, direct and specific activation of either exchange protein directly activated by cAMP (Epac) or protein kinase A (PKA) reduced cigarette smoke extract (CSE)-induced IL-8 mRNA expression and protein release by human ASM cells. CSE-induced IκBα-degradation and p65 nuclear translocation, processes that were primarily reversed by Epac activation. Further, CSE increased extracellular signal-regulated kinase (ERK) phosphorylation, which was selectively reduced by PKA activation. CSE decreased Epac1 expression, but did not affect Epac2 and PKA expression. Importantly, Epac1 expression was also reduced in lung tissue from COPD patients. In conclusion, Epac and PKA decrease CSE-induced IL-8 release by human ASM cells via inhibition of NF-κB and ERK, respectively, pointing at these cAMP effectors as potential targets for anti-inflammatory therapy in COPD. However, cigarette smoke exposure may reduce anti-inflammatory effects of cAMP elevating agents via down-regulation of Epac1.
Conflict of interest statement
Figures
References
-
- Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol. 2009;4:435–459. - PubMed
-
- Quint JK, Wedzicha JA. The neutrophil in chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2007;119:1065–1071. - PubMed
-
- Hurst JR, Perera WR, Wilkinson TM, Donaldson GC, Wedzicha JA. Systemic and upper and lower airway inflammation at exacerbation of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173:71–78. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
