

Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features
- PMID: 22366791
- PMCID: PMC3286330
- DOI: 10.1093/brain/awr361
Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features
Abstract
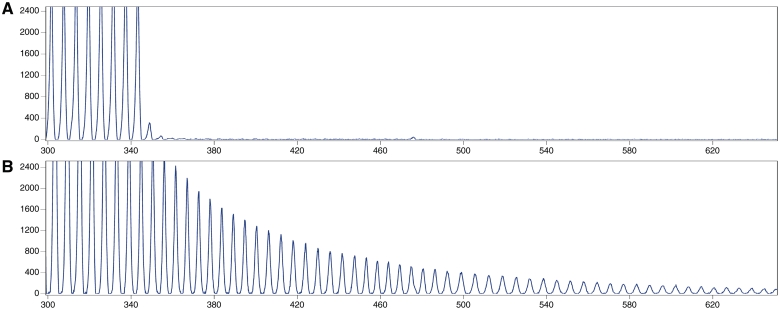
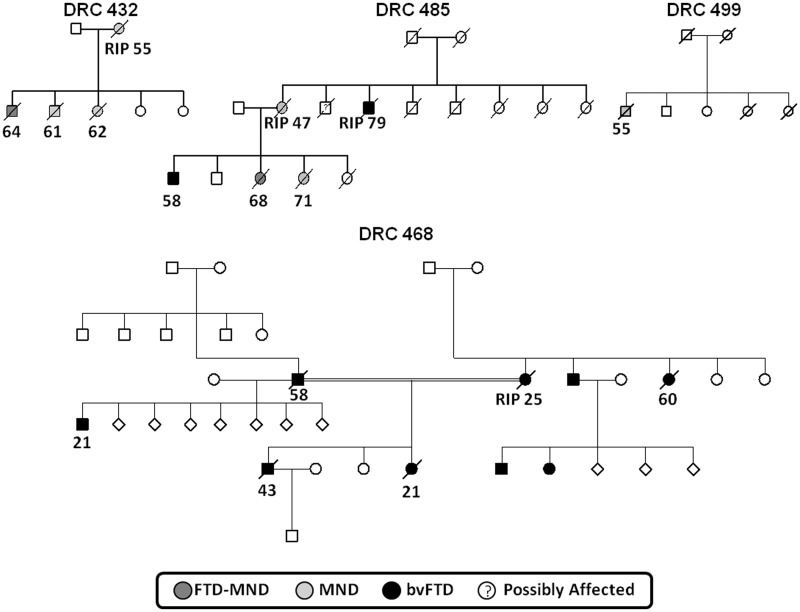
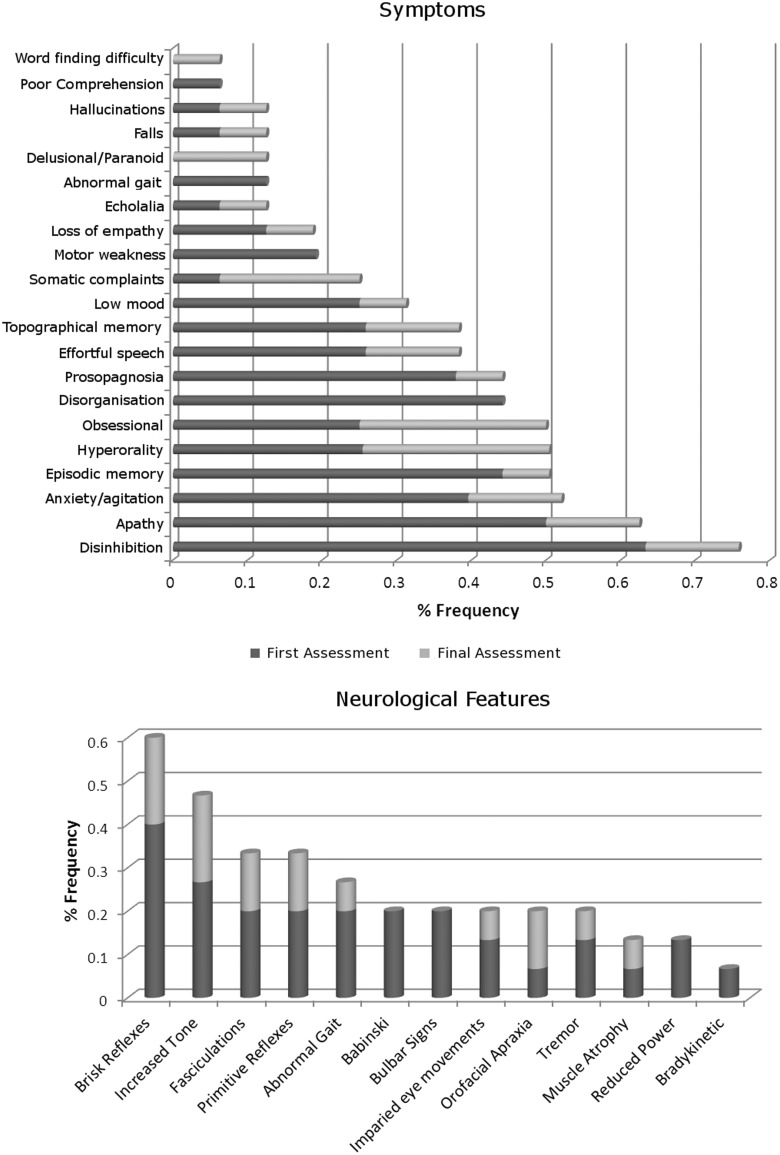
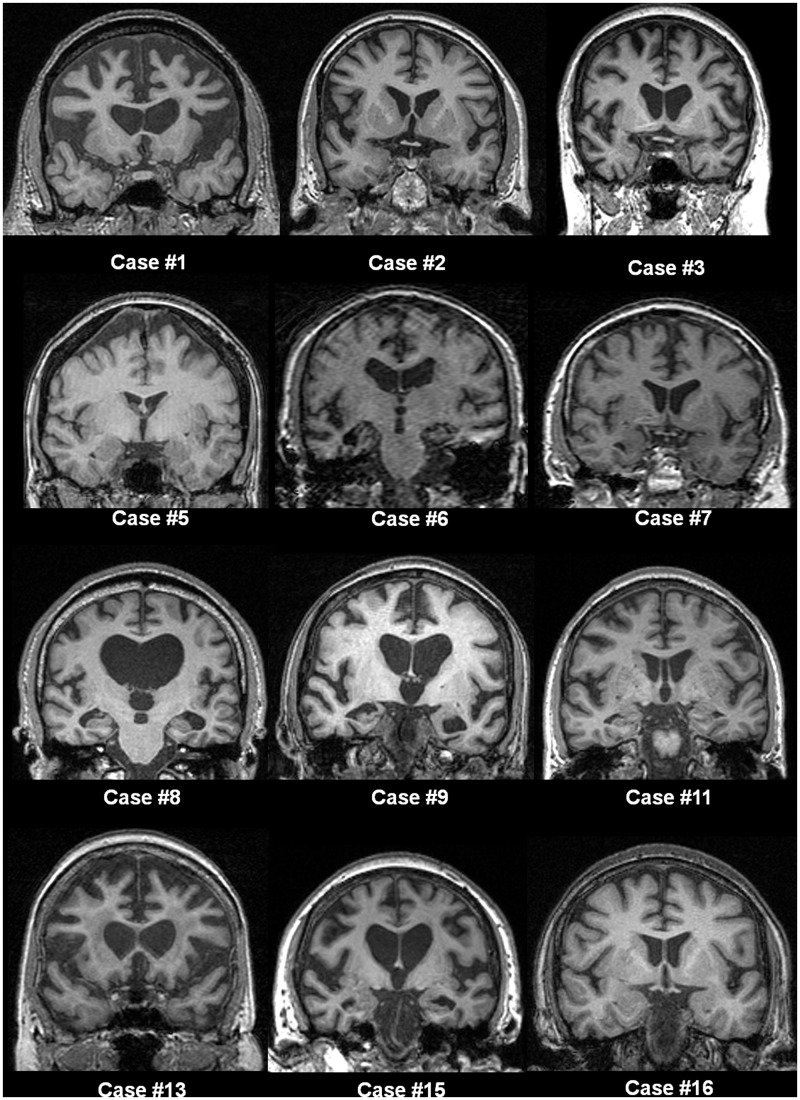
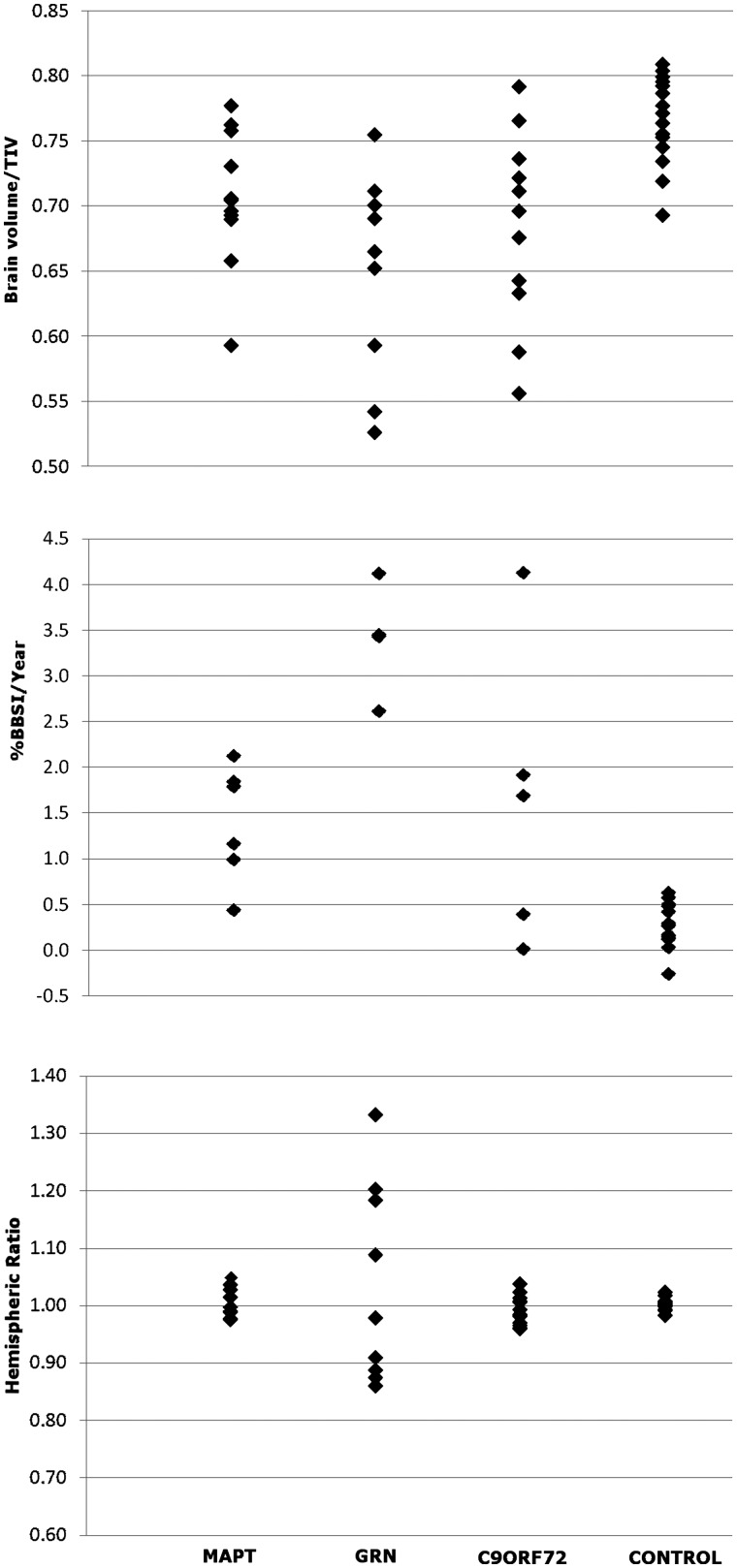
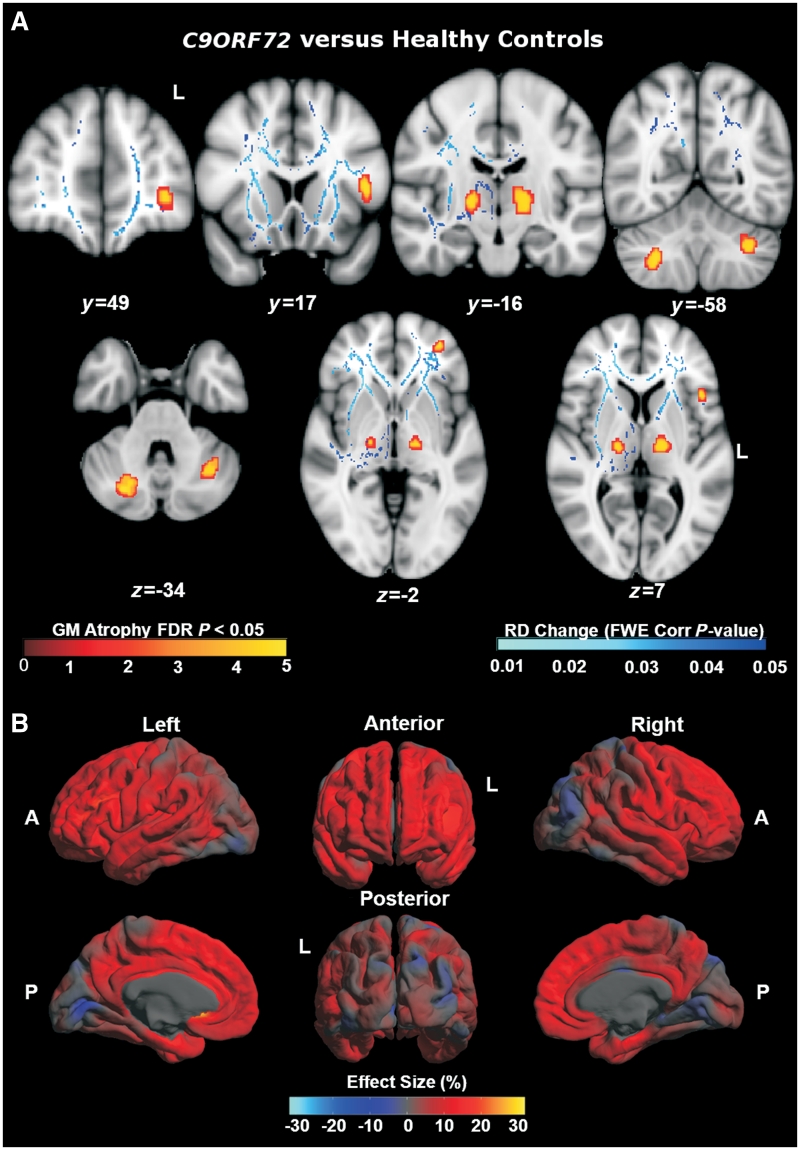
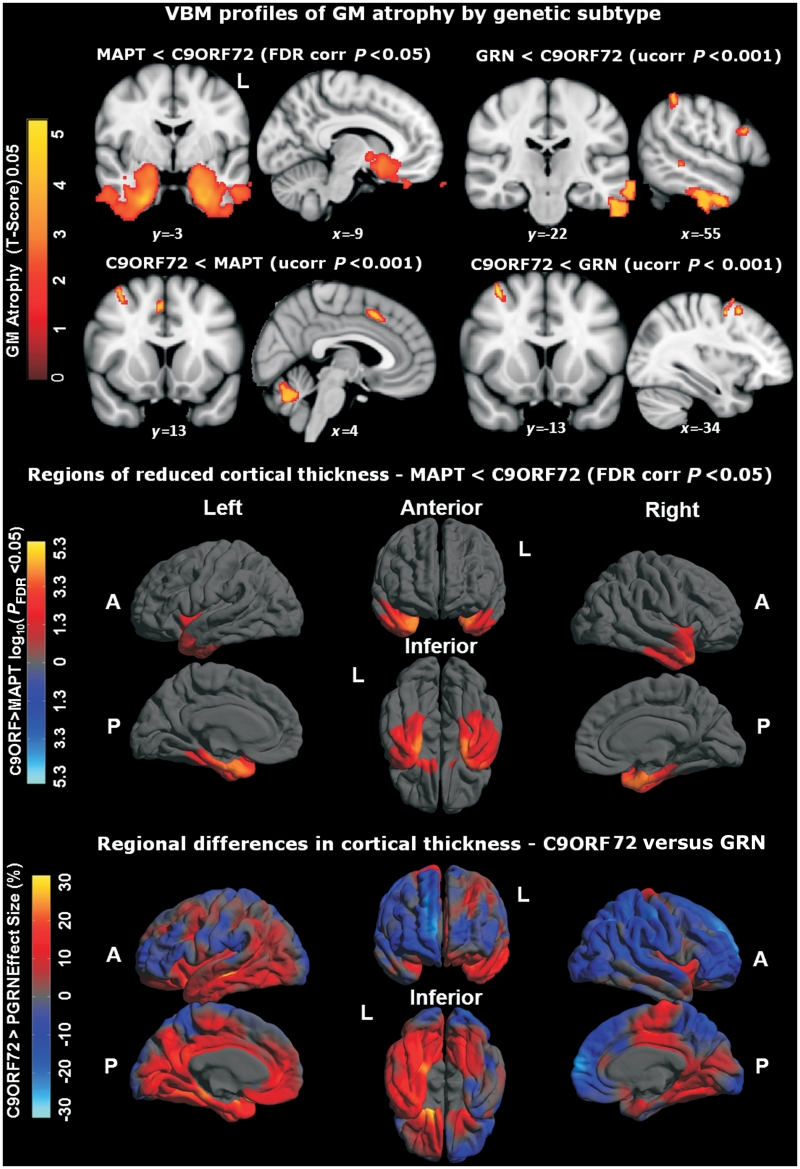
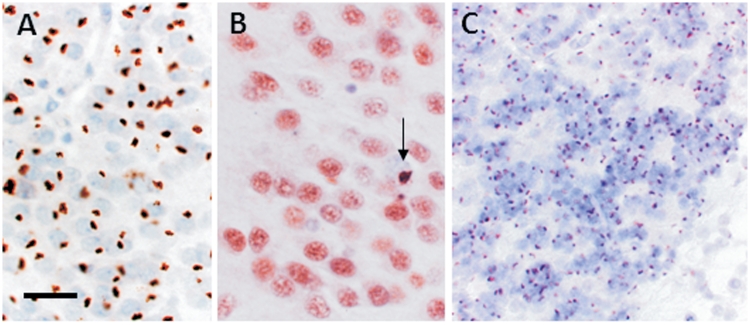
An expanded hexanucleotide repeat in the C9ORF72 gene has recently been identified as a major cause of familial frontotemporal lobar degeneration and motor neuron disease, including cases previously identified as linked to chromosome 9. Here we present a detailed retrospective clinical, neuroimaging and histopathological analysis of a C9ORF72 mutation case series in relation to other forms of genetically determined frontotemporal lobar degeneration ascertained at a specialist centre. Eighteen probands (19 cases in total) were identified, representing 35% of frontotemporal lobar degeneration cases with identified mutations, 36% of cases with clinical evidence of motor neuron disease and 7% of the entire cohort. Thirty-three per cent of these C9ORF72 cases had no identified relevant family history. Families showed wide variation in clinical onset (43-68 years) and duration (1.7-22 years). The most common presenting syndrome (comprising a half of cases) was behavioural variant frontotemporal dementia, however, there was substantial clinical heterogeneity across the C9ORF72 mutation cohort. Sixty per cent of cases developed clinical features consistent with motor neuron disease during the period of follow-up. Anxiety and agitation and memory impairment were prominent features (between a half to two-thirds of cases), and dominant parietal dysfunction was also frequent. Affected individuals showed variable magnetic resonance imaging findings; however, relative to healthy controls, the group as a whole showed extensive thinning of frontal, temporal and parietal cortices, subcortical grey matter atrophy including thalamus and cerebellum and involvement of long intrahemispheric, commissural and corticospinal tracts. The neuroimaging profile of the C9ORF72 expansion was significantly more symmetrical than progranulin mutations with significantly less temporal lobe involvement than microtubule-associated protein tau mutations. Neuropathological examination in six cases with C9ORF72 mutation from the frontotemporal lobar degeneration series identified histomorphological features consistent with either type A or B TAR DNA-binding protein-43 deposition; however, p62-positive (in excess of TAR DNA-binding protein-43 positive) neuronal cytoplasmic inclusions in hippocampus and cerebellum were a consistent feature of these cases, in contrast to the similar frequency of p62 and TAR DNA-binding protein-43 deposition in 53 control cases with frontotemporal lobar degeneration-TAR DNA-binding protein. These findings corroborate the clinical importance of the C9ORF72 mutation in frontotemporal lobar degeneration, delineate phenotypic and neuropathological features that could help to guide genetic testing, and suggest hypotheses for elucidating the neurobiology of a culprit subcortical network.
Figures
References
-
- Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, et al. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS [Internet] Acta Neuropathologica. 2011 Available from: http://www.springerlink.com/index/10.1007/s00401-011-0911-2 (23 November 2011, date last accessed) - DOI - PubMed
-
- Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol. 2003;54(Suppl 5):S15–S19. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
