

Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72
- PMID: 22366793
- PMCID: PMC3286335
- DOI: 10.1093/brain/aws004
Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72
Abstract
Numerous kindreds with familial frontotemporal dementia and/or amyotrophic lateral sclerosis have been linked to chromosome 9, and an expansion of the GGGGCC hexanucleotide repeat in the non-coding region of chromosome 9 open reading frame 72 has recently been identified as the pathogenic mechanism. We describe the key characteristics in the probands and their affected relatives who have been evaluated at Mayo Clinic Rochester or Mayo Clinic Florida in whom the hexanucleotide repeat expansion were found. Forty-three probands and 10 of their affected relatives with DNA available (total 53 subjects) were shown to carry the hexanucleotide repeat expansion. Thirty-six (84%) of the 43 probands had a familial disorder, whereas seven (16%) appeared to be sporadic. Among examined subjects from the 43 families (n = 63), the age of onset ranged from 33 to 72 years (median 52 years) and survival ranged from 1 to 17 years, with the age of onset <40 years in six (10%) and >60 in 19 (30%). Clinical diagnoses among examined subjects included behavioural variant frontotemporal dementia with or without parkinsonism (n = 30), amyotrophic lateral sclerosis (n = 18), frontotemporal dementia/amyotrophic lateral sclerosis with or without parkinsonism (n = 12), and other various syndromes (n = 3). Parkinsonism was present in 35% of examined subjects, all of whom had behavioural variant frontotemporal dementia or frontotemporal dementia/amyotrophic lateral sclerosis as the dominant clinical phenotype. No subject with a diagnosis of primary progressive aphasia was identified with this mutation. Incomplete penetrance was suggested in two kindreds, and the youngest generation had significantly earlier age of onset (>10 years) compared with the next oldest generation in 11 kindreds. Neuropsychological testing showed a profile of slowed processing speed, complex attention/executive dysfunction, and impairment in rapid word retrieval. Neuroimaging studies showed bilateral frontal abnormalities most consistently, with more variable degrees of parietal with or without temporal changes; no case had strikingly focal or asymmetric findings. Neuropathological examination of 14 patients revealed a range of transactive response DNA binding protein molecular weight 43 pathology (10 type A and four type B), as well as ubiquitin-positive cerebellar granular neuron inclusions in all but one case. Motor neuron degeneration was detected in nine patients, including five patients without ante-mortem signs of motor neuron disease. While variability exists, most cases with this mutation have a characteristic spectrum of demographic, clinical, neuropsychological, neuroimaging and especially neuropathological findings.
Figures
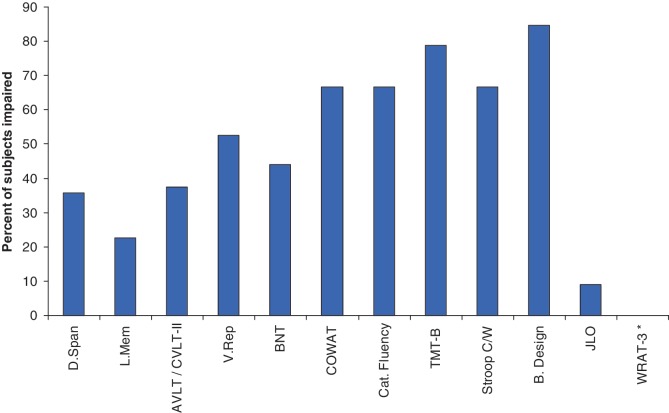
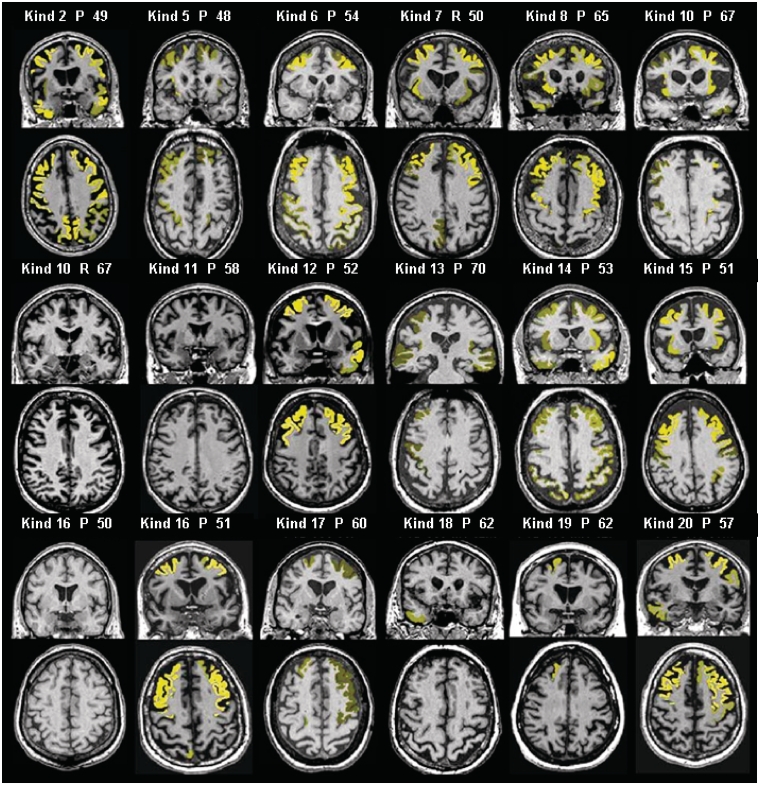



References
-
- Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, et al. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011;122:691–702. - PubMed
-
- Ashburner J, Friston K. Unified segmentation. Neuroimage. 2005;26:839–51. - PubMed
-
- Baker M, Mackenzie I, Pickering-Brown S, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–9. - PubMed
-
- Benton A, Sivan A, Hamsher K, Varney N, Spreen O. Contributions to neuropsychological assessment: A clinical manual. second edn. New York: Oxford University Press; 1994.
-
- Bornstein R. Normative data on selected neuropsychological measures from a nonclinical sample. J Clin Psychol. 1985;42:651–9.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
