

Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72
- PMID: 22366794
- PMCID: PMC3286333
- DOI: 10.1093/brain/awr366
Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72
Abstract
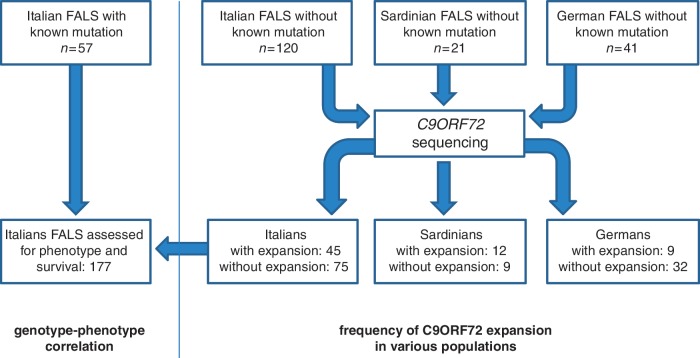
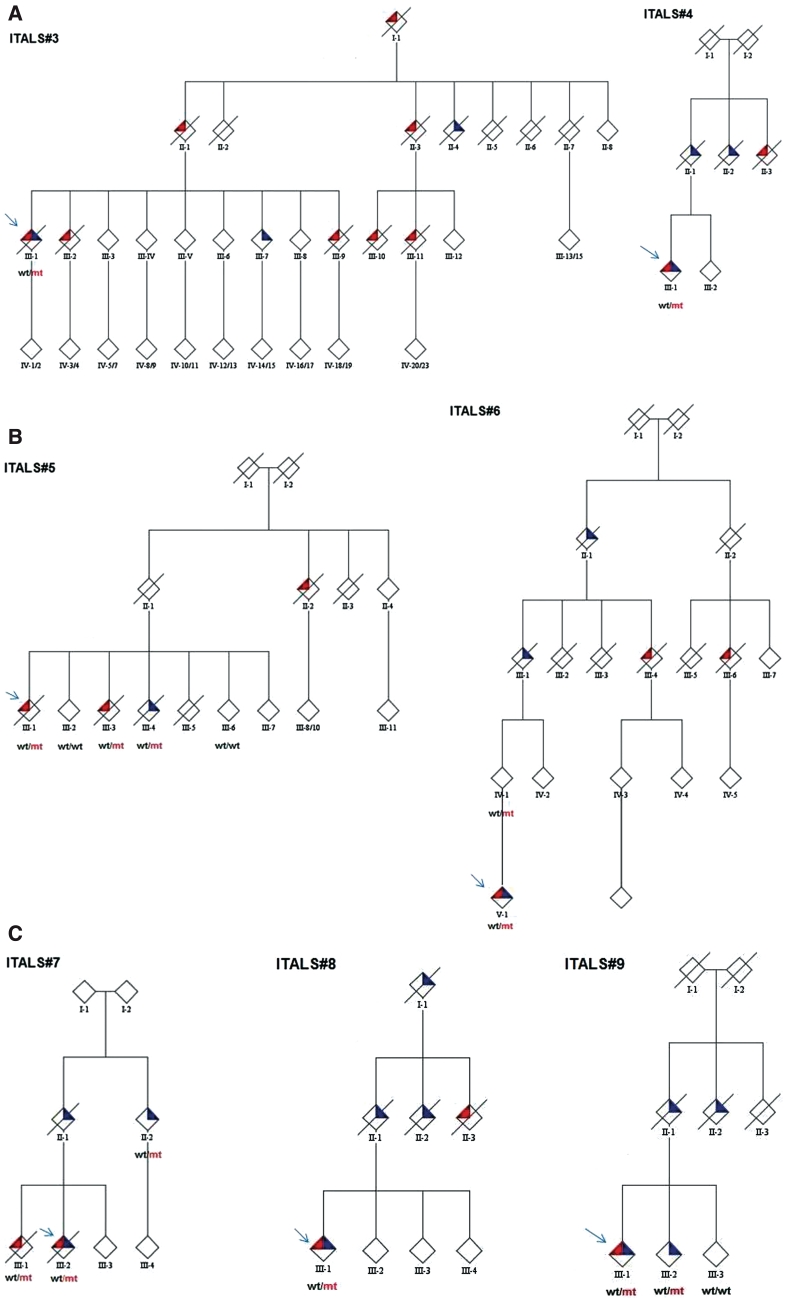
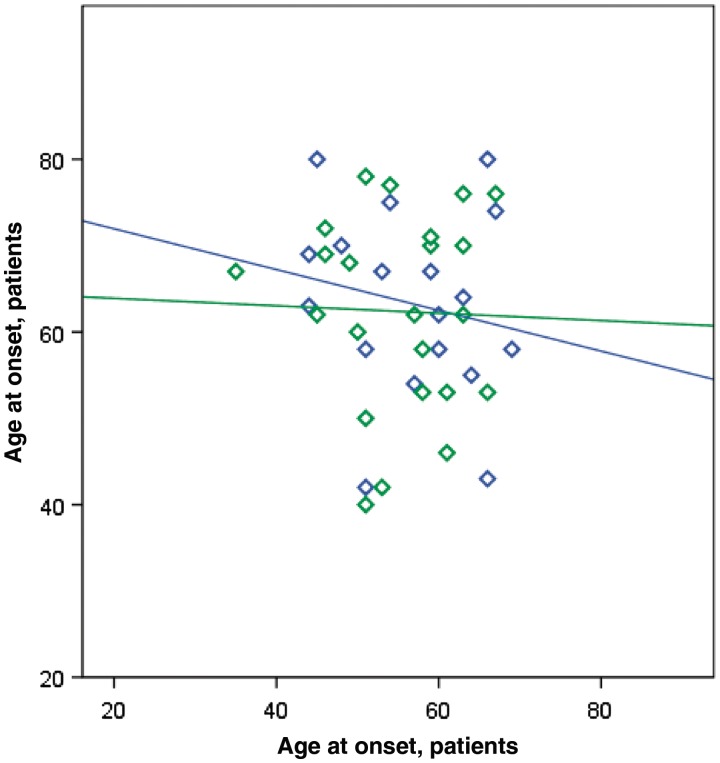
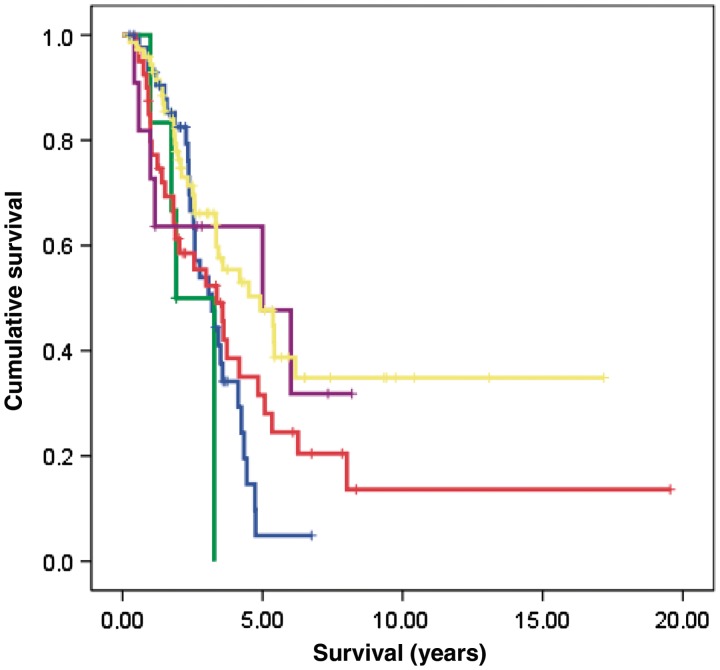
A large hexanucleotide (GGGGCC) repeat expansion in the first intron of C9ORF72, a gene located on chromosome 9p21, has been recently reported to be responsible for ~40% of familial amyotrophic lateral sclerosis cases of European ancestry. The aim of the current article was to describe the phenotype of amyotrophic lateral sclerosis cases carrying the expansion by providing a detailed clinical description of affected cases from representative multi-generational kindreds, and by analysing the age of onset, gender ratio and survival in a large cohort of patients with familial amyotrophic lateral sclerosis. We collected DNA and analysed phenotype data for 141 index Italian familial amyotrophic lateral sclerosis cases (21 of Sardinian ancestry) and 41 German index familial amyotrophic lateral sclerosis cases. Pathogenic repeat expansions were detected in 45 (37.5%) patients from mainland Italy, 12 (57.1%) patients of Sardinian ancestry and nine (22.0%) of the 41 German index familial amyotrophic lateral sclerosis cases. The disease was maternally transmitted in 27 (49.1%) pedigrees and paternally transmitted in 28 (50.9%) pedigrees (P = non-significant). On average, children developed disease 7.0 years earlier than their parents [children: 55.8 years (standard deviation 7.9), parents: 62.8 (standard deviation 10.9); P = 0.003]. Parental phenotype influenced the type of clinical symptoms manifested by the child: of the 13 cases where the affected parent had an amyotrophic lateral sclerosis-frontotemporal dementia or frontotemporal dementia, the affected child also developed amyotrophic lateral sclerosis-frontotemporal dementia in nine cases. When compared with patients carrying mutations of other amyotrophic lateral sclerosis-related genes, those with C9ORF72 expansion had commonly a bulbar onset (42.2% compared with 25.0% among non-C9ORF72 expansion cases, P = 0.03) and cognitive impairment (46.7% compared with 9.1% among non-C9ORF72 expansion cases, P = 0.0001). Median survival from symptom onset among cases carrying C9ORF72 repeat expansion was 3.2 years lower than that of patients carrying TARDBP mutations (5.0 years; 95% confidence interval: 3.6-7.2) and longer than those with FUS mutations (1.9 years; 95% confidence interval: 1.7-2.1). We conclude that C9ORF72 hexanucleotide repeat expansions were the most frequent mutation in our large cohort of patients with familial amyotrophic lateral sclerosis of Italian, Sardinian and German ancestry. Together with mutation of SOD1, TARDBP and FUS, mutations of C9ORF72 account for ~60% of familial amyotrophic lateral sclerosis in Italy. Patients with C9ORF72 hexanucleotide repeat expansions present some phenotypic differences compared with patients with mutations of other genes or with unknown mutations, namely a high incidence of bulbar-onset disease and comorbidity with frontotemporal dementia. Their pedigrees typically display a high frequency of cases with pure frontotemporal dementia, widening the concept of familial amyotrophic lateral sclerosis.
Figures
References
-
- Andersen PM, Abrahams S, Borasio GD, de Carvalho M, Chio A, Van Damme P, et al. EFNS Task Force on Diagnosis and Management of Amyotrophic Lateral Sclerosis. EFNS guidelines on the Clinical Management of Amyotrophic Lateral Sclerosis (MALS) - revised report of an EFNS task force. Eur J Neurol. 2011 doi:10.1111/j.1468-1331.2011.03501.x. - PubMed
-
- Borroni B, Archetti S, Del Bo R, Papetti A, Buratti E, Bonvicini C, et al. TARDBP mutations in frontotemporal lobar degeneration: frequency, clinical features, and disease course. Rejuvenation Res. 2010;13:509–17. - PubMed
-
- Byrne S, Bede P, Elamin M, Kenna K, Lynch C, McLaughlin R, et al. Proposed criteria for familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2011;12:157–9. - PubMed
