

Differential degradation of full-length and cleaved ataxin-7 fragments in a novel stable inducible SCA7 model
- PMID: 22367614
- PMCID: PMC3360856
- DOI: 10.1007/s12031-012-9722-8
Differential degradation of full-length and cleaved ataxin-7 fragments in a novel stable inducible SCA7 model
Abstract
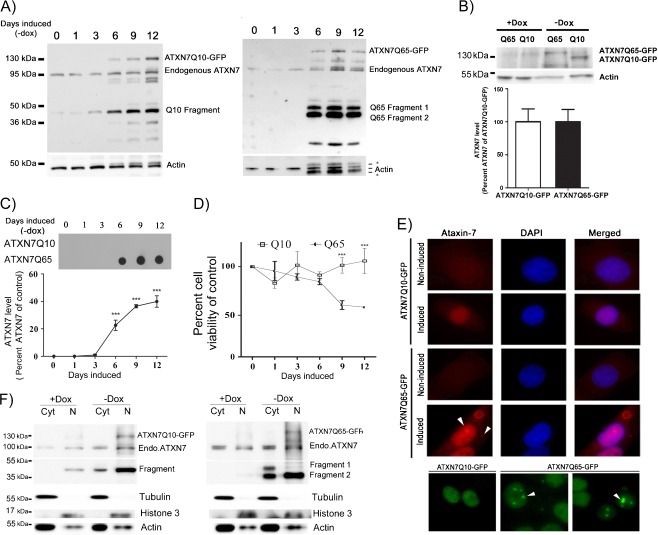
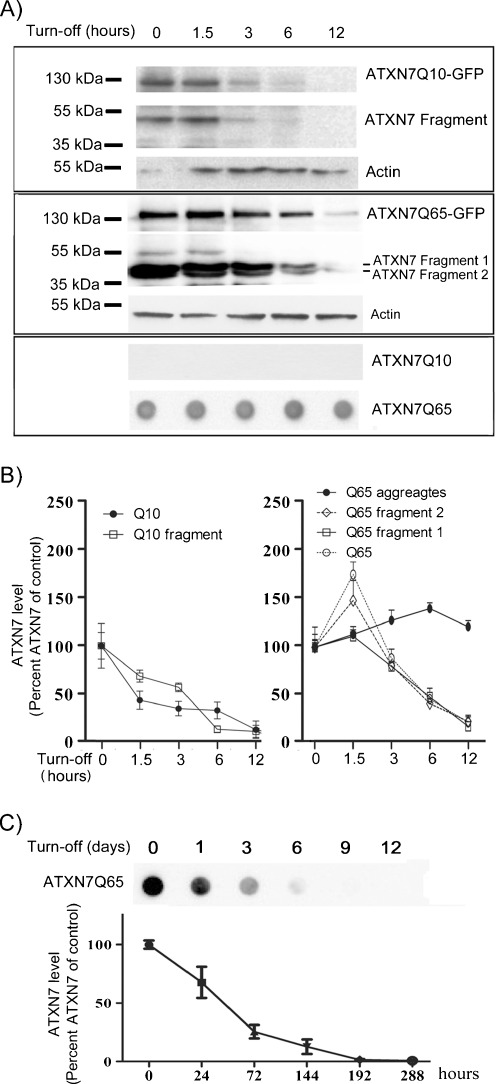
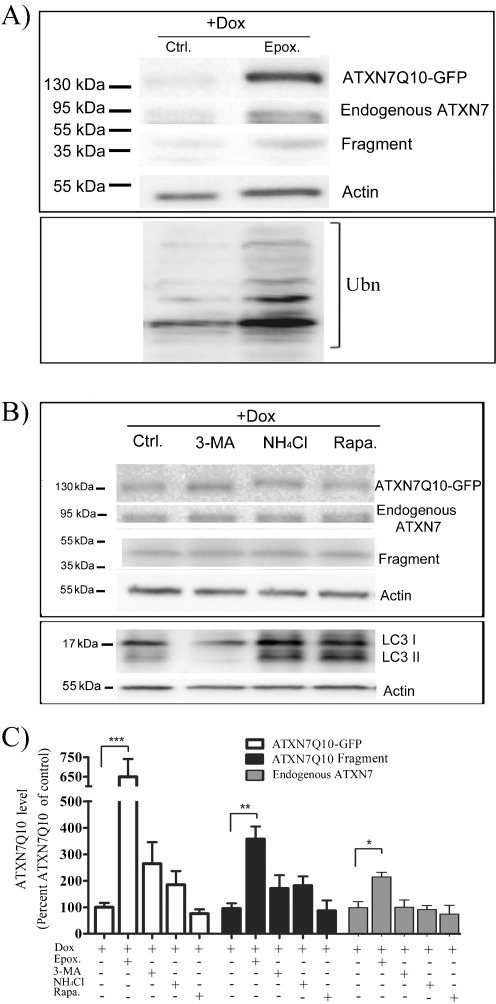
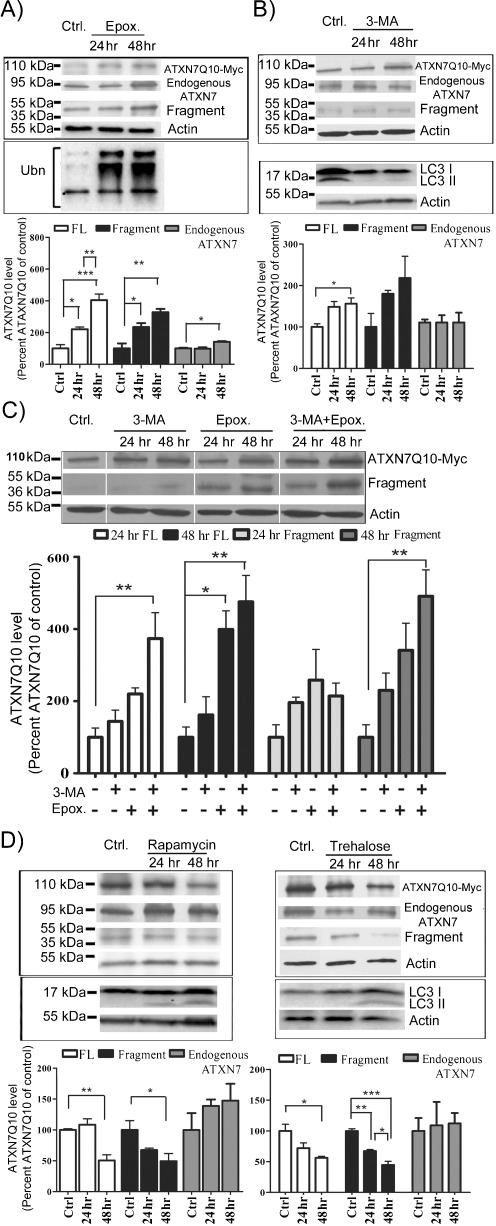
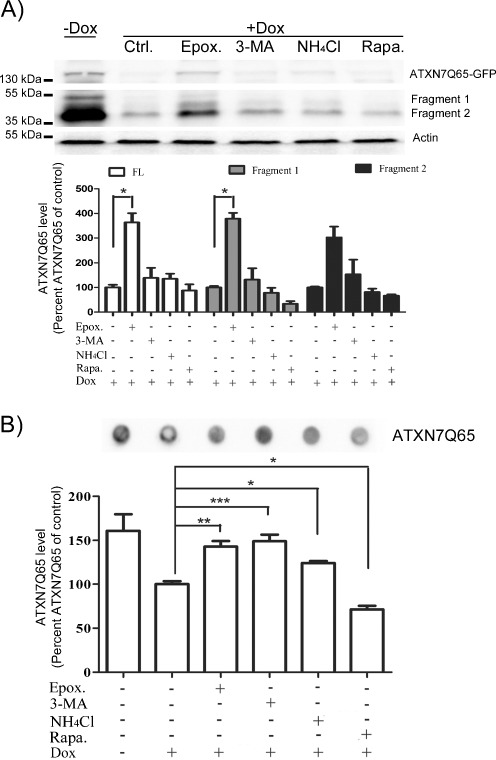
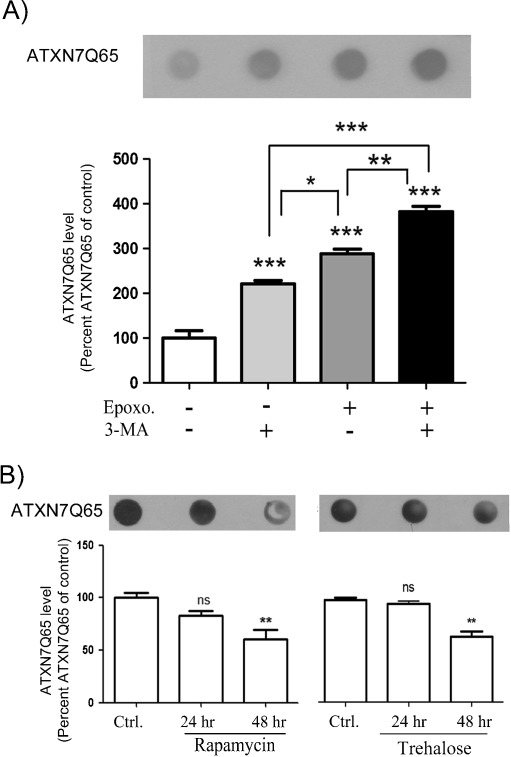
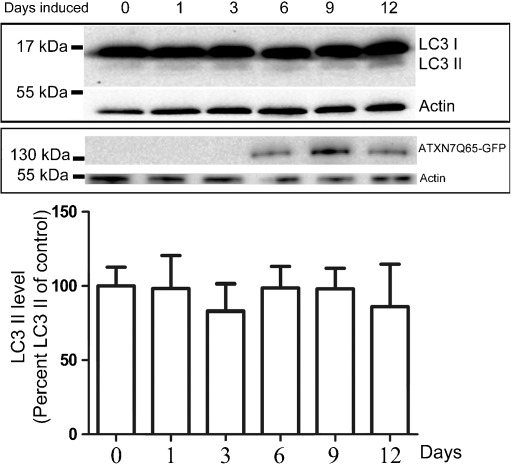
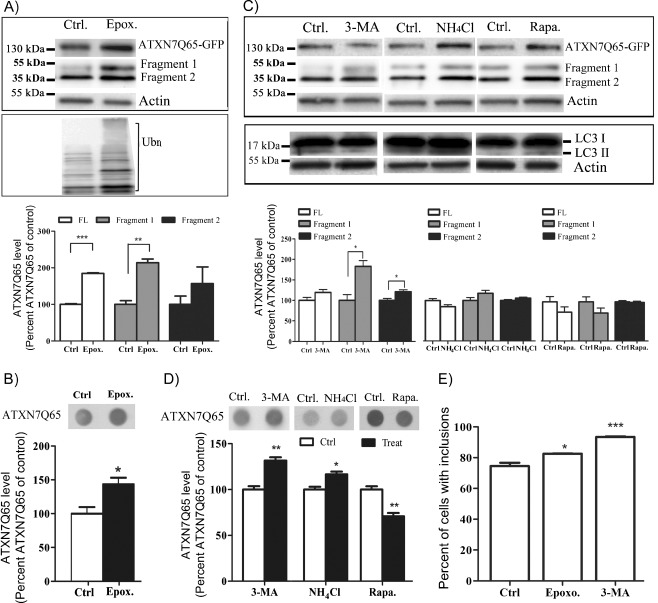
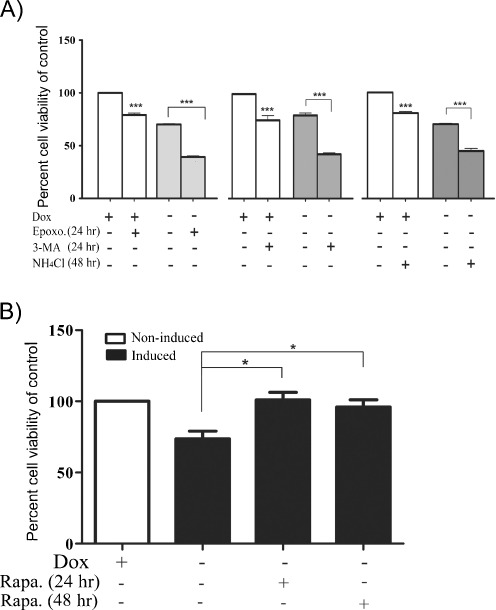
Spinocerebellar ataxia type 7 (SCA7) is one of nine neurodegenerative disorders caused by expanded polyglutamine repeats, and a common toxic gain-of-function mechanism has been proposed. Proteolytic cleavage of several polyglutamine proteins has been identified and suggested to modulate the polyglutamine toxicity. In this study, we show that full-length and cleaved fragments of the SCA7 disease protein ataxin-7 (ATXN7) are differentially degraded. We found that the ubiquitin-proteosome system (UPS) was essential for the degradation of full-length endogenous ATXN7 or transgenic full-length ATXN7 with a normal or expanded glutamine repeat in both HEK 293T and stable PC12 cells. However, a similar contribution by UPS and autophagy was found for the degradation of proteolytically cleaved ATXN7 fragments. Furthermore, in our novel stable inducible PC12 model, induction of mutant ATXN7 expression resulted in toxicity and this toxicity was worsened by inhibition of either UPS or autophagy. In contrast, pharmacological activation of autophagy could ameliorate the ATXN7-induced toxicity. Based on our findings, we propose that both UPS and autophagy are important for the reduction of mutant ataxin-7-induced toxicity, and enhancing ATXN7 clearance through autophagy could be used as a potential therapeutic strategy in SCA7.
Figures
Similar articles
-
Inhibition of autophagy via p53-mediated disruption of ULK1 in a SCA7 polyglutamine disease model.J Mol Neurosci. 2013 Jul;50(3):586-99. doi: 10.1007/s12031-013-0012-x. Epub 2013 Apr 18. J Mol Neurosci. 2013. PMID: 23592174
-
Altered p53 and NOX1 activity cause bioenergetic defects in a SCA7 polyglutamine disease model.Biochim Biophys Acta. 2015 Apr-May;1847(4-5):418-428. doi: 10.1016/j.bbabio.2015.01.012. Epub 2015 Jan 31. Biochim Biophys Acta. 2015. PMID: 25647692
-
Reelin is a target of polyglutamine expanded ataxin-7 in human spinocerebellar ataxia type 7 (SCA7) astrocytes.Proc Natl Acad Sci U S A. 2012 Dec 26;109(52):21319-24. doi: 10.1073/pnas.1218331110. Epub 2012 Dec 10. Proc Natl Acad Sci U S A. 2012. PMID: 23236151 Free PMC article.
-
Molecular Targets and Therapeutic Strategies in Spinocerebellar Ataxia Type 7.Neurotherapeutics. 2019 Oct;16(4):1074-1096. doi: 10.1007/s13311-019-00778-5. Neurotherapeutics. 2019. PMID: 31432449 Free PMC article. Review.
-
Molecular pathogenesis and cellular pathology of spinocerebellar ataxia type 7 neurodegeneration.Cerebellum. 2008;7(2):138-49. doi: 10.1007/s12311-008-0027-y. Cerebellum. 2008. PMID: 18418675 Free PMC article. Review.
Cited by
-
Oxidative Stress in DNA Repeat Expansion Disorders: A Focus on NRF2 Signaling Involvement.Biomolecules. 2020 May 1;10(5):702. doi: 10.3390/biom10050702. Biomolecules. 2020. PMID: 32369911 Free PMC article. Review.
-
Expanded ataxin-7 cause toxicity by inducing ROS production from NADPH oxidase complexes in a stable inducible Spinocerebellar ataxia type 7 (SCA7) model.BMC Neurosci. 2012 Jul 24;13:86. doi: 10.1186/1471-2202-13-86. BMC Neurosci. 2012. PMID: 22827889 Free PMC article.
-
Development of a Polymeric Pharmacological Nanocarrier System as a Potential Therapy for Spinocerebellar Ataxia Type 7.Cells. 2023 Nov 30;12(23):2735. doi: 10.3390/cells12232735. Cells. 2023. PMID: 38067163 Free PMC article.
-
Motor Performances of Spontaneous and Genetically Modified Mutants with Cerebellar Atrophy.Cerebellum. 2019 Jun;18(3):615-634. doi: 10.1007/s12311-019-01017-5. Cerebellum. 2019. PMID: 30820866 Review.
-
From pathways to targets: understanding the mechanisms behind polyglutamine disease.Biomed Res Int. 2014;2014:701758. doi: 10.1155/2014/701758. Epub 2014 Sep 21. Biomed Res Int. 2014. PMID: 25309920 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
