

SHP-1 as a critical regulator of Mycoplasma pneumoniae-induced inflammation in human asthmatic airway epithelial cells
- PMID: 22371396
- PMCID: PMC3880785
- DOI: 10.4049/jimmunol.1100573
SHP-1 as a critical regulator of Mycoplasma pneumoniae-induced inflammation in human asthmatic airway epithelial cells
Abstract
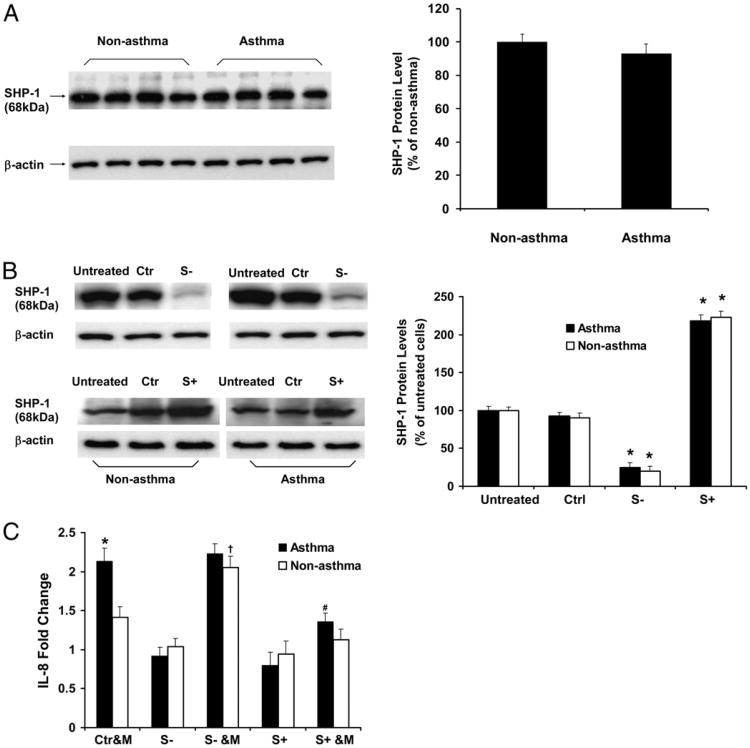
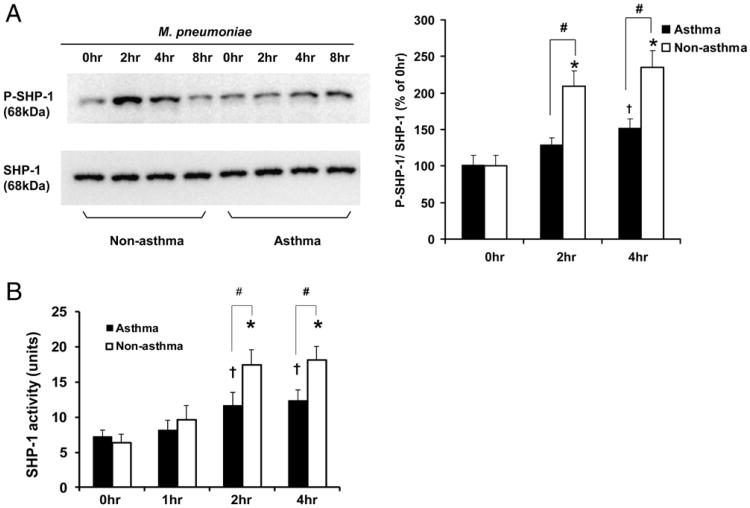
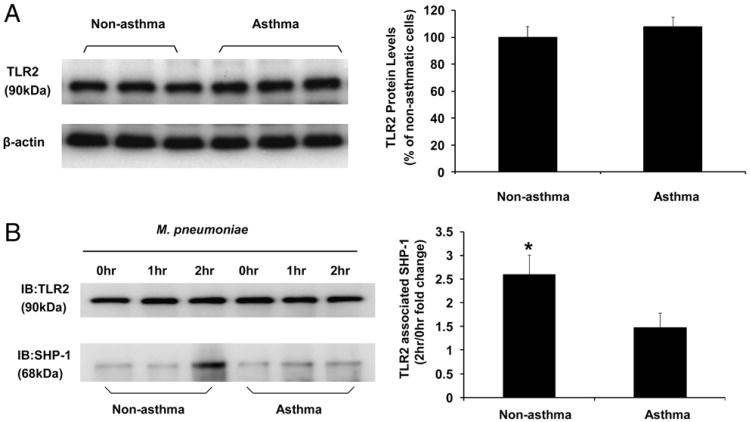
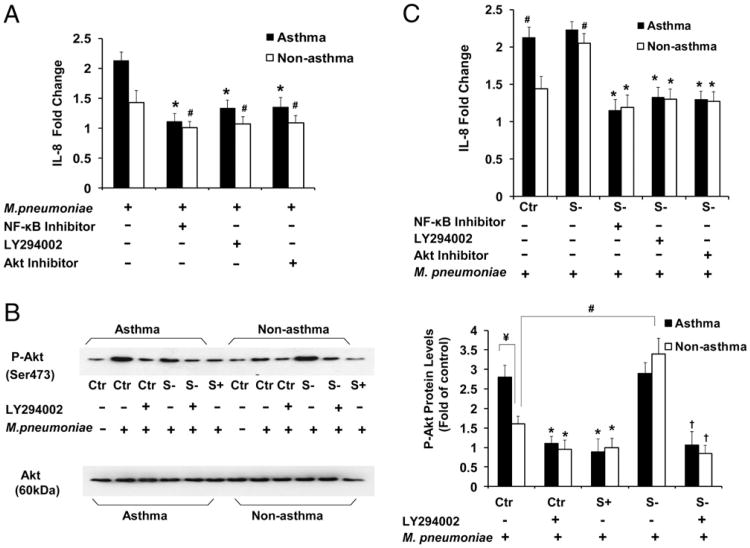
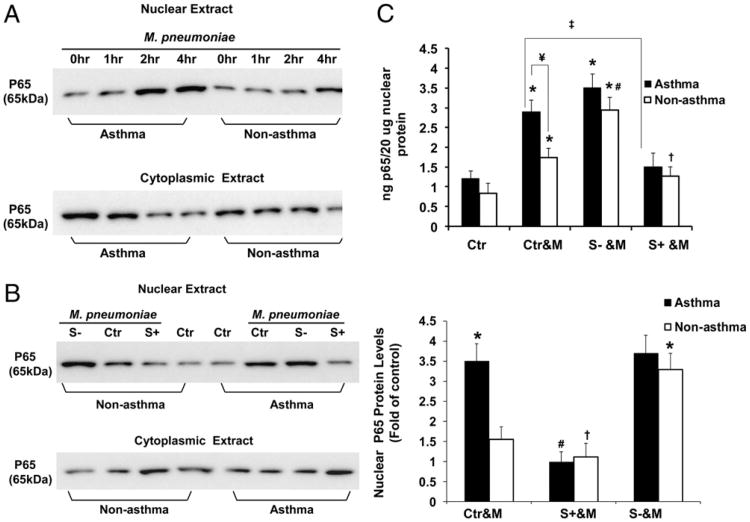
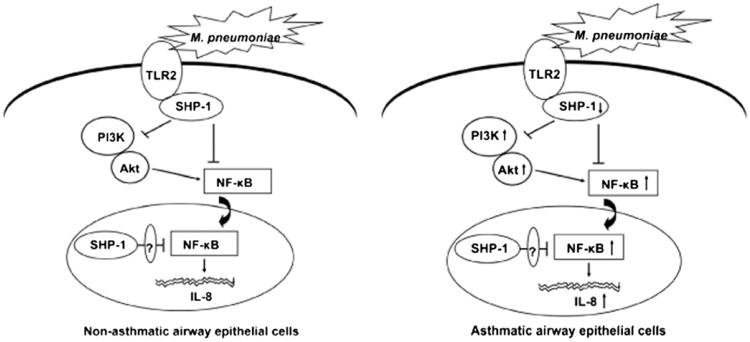
Asthma is a chronic inflammatory disease in which airway epithelial cells are the first line of defense against exposure of the airway to infectious agents. Src homology protein (SHP)-1, a protein tyrosine phosphatase, is a negative regulator of signaling pathways that are critical to the development of asthma and host defense. We hypothesize that SHP-1 function is defective in asthma, contributing to the increased inflammatory response induced by Mycoplasma pneumoniae, a pathogen known to exacerbate asthma. M. pneumoniae significantly activated SHP-1 in airway epithelial cells collected from nonasthmatic subjects by bronchoscopy with airway brushing but not in cells from asthmatic subjects. In asthmatic airway epithelial cells, M. pneumoniae induced significant PI3K/Akt phosphorylation, NF-κB activation, and IL-8 production compared with nonasthmatic cells, which were reversed by SHP-1 overexpression. Conversely, SHP-1 knockdown significantly increased IL-8 production and PI3K/Akt and NF-κB activation in the setting of M. pneumoniae infection in nonasthmatic cells, but it did not exacerbate these three parameters already activated in asthmatic cells. Thus, SHP-1 plays a critical role in abrogating M. pneumoniae-induced IL-8 production in nonasthmatic airway epithelial cells through inhibition of PI3K/Akt and NF-κB activity, but it is defective in asthma, resulting in an enhanced inflammatory response to infection.
Figures
Similar articles
-
Surfactant protein A is defective in abrogating inflammation in asthma.Am J Physiol Lung Cell Mol Physiol. 2011 Oct;301(4):L598-606. doi: 10.1152/ajplung.00381.2010. Epub 2011 Jul 22. Am J Physiol Lung Cell Mol Physiol. 2011. PMID: 21784968 Free PMC article.
-
Distinct domains in the SHP-2 phosphatase differentially regulate epidermal growth factor receptor/NF-kappaB activation through Gab1 in glioblastoma cells.Mol Cell Biol. 2004 Jan;24(2):823-36. doi: 10.1128/MCB.24.2.823-836.2004. Mol Cell Biol. 2004. PMID: 14701753 Free PMC article.
-
Modulation of IL-8 boosted by Mycoplasma pneumoniae lysate in human airway epithelial cells.J Clin Immunol. 2013 Aug;33(6):1117-25. doi: 10.1007/s10875-013-9909-y. Epub 2013 Jun 19. J Clin Immunol. 2013. PMID: 23779254
-
Interleukin 1 Receptor-Like 1 (IL1RL1) Promotes Airway Bacterial and Viral Infection and Inflammation.Infect Immun. 2019 Jun 20;87(7):e00340-19. doi: 10.1128/IAI.00340-19. Print 2019 Jul. Infect Immun. 2019. PMID: 31061143 Free PMC article.
-
Tyrosine phosphatase SHP-1 in oxidative stress and development of allergic airway inflammation.Am J Respir Cell Mol Biol. 2008 Oct;39(4):412-9. doi: 10.1165/rcmb.2007-0229OC. Epub 2008 Apr 25. Am J Respir Cell Mol Biol. 2008. PMID: 18441283 Free PMC article.
Cited by
-
Protein tyrosine phosphatase SHP-1: resurgence as new drug target for human autoimmune disorders.Immunol Res. 2016 Aug;64(4):804-19. doi: 10.1007/s12026-016-8805-y. Immunol Res. 2016. PMID: 27216862 Review.
-
Macrophage-activating lipopeptide-2 requires Mal and PI3K for efficient induction of heme oxygenase-1.PLoS One. 2014 Jul 31;9(7):e103433. doi: 10.1371/journal.pone.0103433. eCollection 2014. PLoS One. 2014. PMID: 25077631 Free PMC article.
-
Modulation of IL-4/IL-13 cytokine signaling in the context of allergic disease.J Allergy Clin Immunol. 2022 Aug;150(2):266-276. doi: 10.1016/j.jaci.2022.06.012. J Allergy Clin Immunol. 2022. PMID: 35934680 Free PMC article. Review.
-
HNRNPH1 Is a Novel Regulator Of Cellular Proliferation and Disease Progression in Chronic Myeloid Leukemia.Front Oncol. 2021 Jul 6;11:682859. doi: 10.3389/fonc.2021.682859. eCollection 2021. Front Oncol. 2021. PMID: 34295818 Free PMC article.
-
A computational-based new treatment strategy with three-armed RCT on Mycoplasma pneumoniae pneumonia in children.Chin Med. 2025 Jul 1;20(1):97. doi: 10.1186/s13020-025-01149-3. Chin Med. 2025. PMID: 40597150 Free PMC article.
References
-
- Djukanović R, Wilson JW, Britten KM, Wilson SJ, Walls AF, Roche WR, Howarth PH, Holgate ST. Effect of an inhaled corticosteroid on airway inflammation and symptoms in asthma. Am Rev Respir Dis. 1992;145:669–674. - PubMed
-
- Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–2261. - PubMed
-
- Allegra L, Blasi F, Centanni S, Cosentini R, Denti F, Raccanelli R, Tarsia P, Valenti V. Acute exacerbations of asthma in adults: role of Chlamydia pneumoniae infection. Eur Respir J. 1994;7:2165–2168. - PubMed
-
- Berkovich S, Millian SJ, Snyder RD. The association of viral and mycoplasma infections with recurrence of wheezing in the asthmatic child. Ann Allergy. 1970;28:43–49. - PubMed
-
- Hahn DL, Dodge RW, Golubjatnikov R. Association of Chlamydia pneumoniae (strain TWAR) infection with wheezing, asthmatic bronchitis, and adult-onset asthma. JAMA. 1991;266:225–230. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
