

Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice
- PMID: 22371574
- PMCID: PMC3306692
- DOI: 10.1073/pnas.1119908109
Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice
Abstract
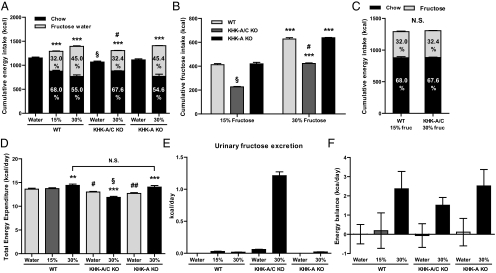
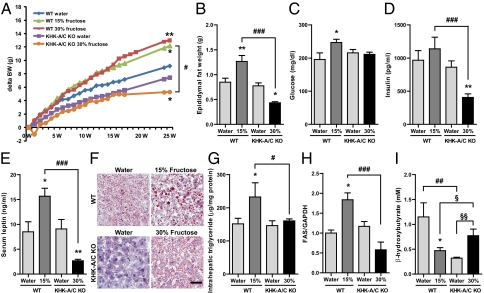
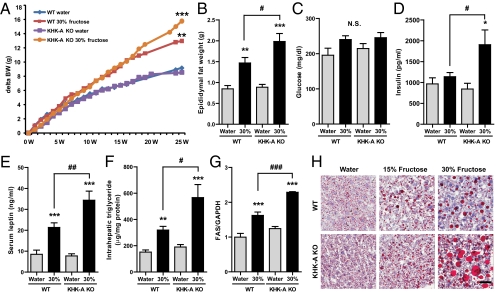
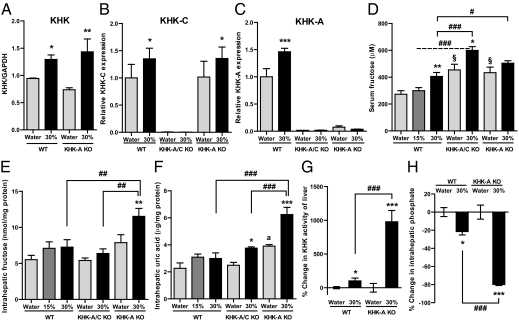
Fructose intake from added sugars correlates with the epidemic rise in obesity, metabolic syndrome, and nonalcoholic fatty liver disease. Fructose intake also causes features of metabolic syndrome in laboratory animals and humans. The first enzyme in fructose metabolism is fructokinase, which exists as two isoforms, A and C. Here we show that fructose-induced metabolic syndrome is prevented in mice lacking both isoforms but is exacerbated in mice lacking fructokinase A. Fructokinase C is expressed primarily in liver, intestine, and kidney and has high affinity for fructose, resulting in rapid metabolism and marked ATP depletion. In contrast, fructokinase A is widely distributed, has low affinity for fructose, and has less dramatic effects on ATP levels. By reducing the amount of fructose for metabolism in the liver, fructokinase A protects against fructokinase C-mediated metabolic syndrome. These studies provide insights into the mechanisms by which fructose causes obesity and metabolic syndrome.
Conflict of interest statement
Conflict of interest statement: Based on the discoveries from this study, T.I., M.A.L., and R.J.J. are listed as inventors on a patent application from the University of Colorado related to developing isoform-specific fructokinase inhibitors in the treatment of disorders associated with obesity and insulin resistance. No other authors have any conflicts of interest.
Figures

Comment in
-
Opposite fates of fructose in the development of metabolic syndrome.World J Gastroenterol. 2012 Sep 7;18(33):4478-80. doi: 10.3748/wjg.v18.i33.4478. World J Gastroenterol. 2012. PMID: 22969219 Free PMC article.
References
-
- Havel PJ. Dietary fructose: Implications for dysregulation of energy homeostasis and lipid/carbohydrate metabolism. Nutr Rev. 2005;63:133–157. - PubMed
-
- Tappy L, Lê KA. Metabolic effects of fructose and the worldwide increase in obesity. Physiol Rev. 2010;90:23–46. - PubMed
-
- Johnson RJ, et al. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am J Clin Nutr. 2007;86:899–906. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
