

ClipCrop: a tool for detecting structural variations with single-base resolution using soft-clipping information
- PMID: 22373054
- PMCID: PMC3287472
- DOI: 10.1186/1471-2105-12-S14-S7
ClipCrop: a tool for detecting structural variations with single-base resolution using soft-clipping information
Abstract
Background: Structural variations (SVs) change the structure of the genome and are therefore the causes of various diseases. Next-generation sequencing allows us to obtain a multitude of sequence data, some of which can be used to infer the position of SVs.
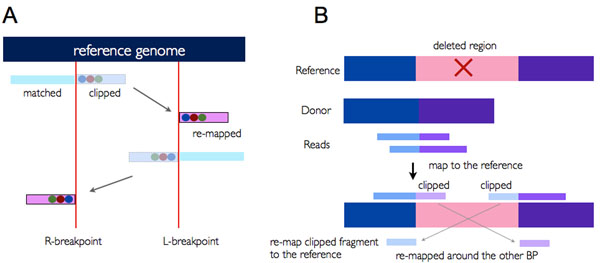
Methods: We developed a new method and implementation named ClipCrop for detecting SVs with single-base resolution using soft-clipping information. A soft-clipped sequence is an unmatched fragment in a partially mapped read. To assess the performance of ClipCrop with other SV-detecting tools, we generated various patterns of simulation data - SV lengths, read lengths, and the depth of coverage of short reads - with insertions, deletions, tandem duplications, inversions and single nucleotide alterations in a human chromosome. For comparison, we selected BreakDancer, CNVnator and Pindel, each of which adopts a different approach to detect SVs, e.g. discordant pair approach, depth of coverage approach and split read approach, respectively.
Results: Our method outperformed BreakDancer and CNVnator in both discovering rate and call accuracy in any type of SV. Pindel offered a similar performance as our method, but our method crucially outperformed for detecting small duplications. From our experiments, ClipCrop infer reliable SVs for the data set with more than 50 bases read lengths and 20x depth of coverage, both of which are reasonable values in current NGS data set.
Conclusions: ClipCrop can detect SVs with higher discovering rate and call accuracy than any other tool in our simulation data set.
Figures







References
-
- McCarroll Steven A, Altshuler David M. Copy-number variation and association studies of human disease. Nat. Genetics. 2009;39:S37–S42. - PubMed
-
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, Zhang R, Lee YH, Hicks J, Spence SJ, Lee AT, Puura K, Lehtimäki T, Ledbetter D, Gregersen PK, Bregman J, Sutcliffe JS, Jobanputra V, Chung W, Warburton D, King MC, Skuse D, Geschwind DH, Gilliam TC, Ye K, Wigler M. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. - DOI - PMC - PubMed
-
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. Alpha-synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. - DOI - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
