

Comprehensive evaluation of matrix factorization methods for the analysis of DNA microarray gene expression data
- PMID: 22373334
- PMCID: PMC3278848
- DOI: 10.1186/1471-2105-12-S13-S8
Comprehensive evaluation of matrix factorization methods for the analysis of DNA microarray gene expression data
Abstract
Background: Clustering-based methods on gene-expression analysis have been shown to be useful in biomedical applications such as cancer subtype discovery. Among them, Matrix factorization (MF) is advantageous for clustering gene expression patterns from DNA microarray experiments, as it efficiently reduces the dimension of gene expression data. Although several MF methods have been proposed for clustering gene expression patterns, a systematic evaluation has not been reported yet.
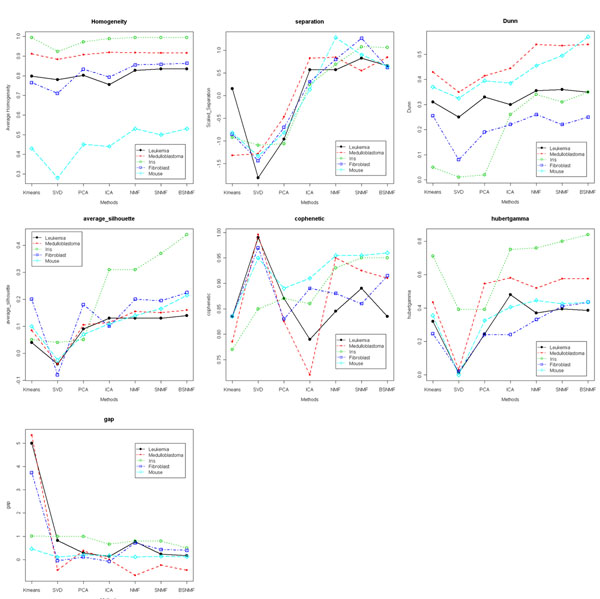
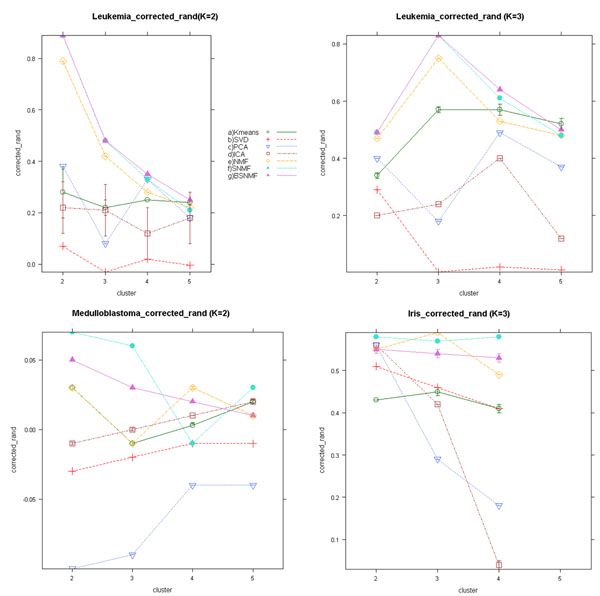
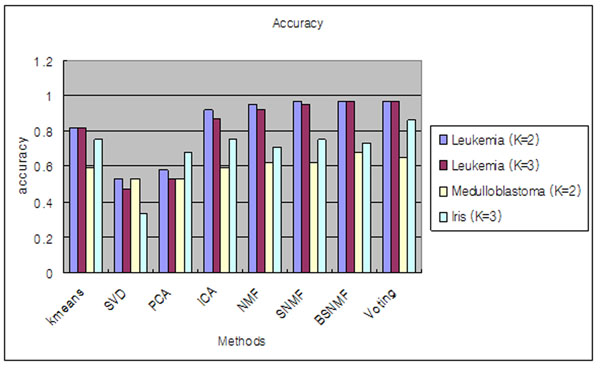
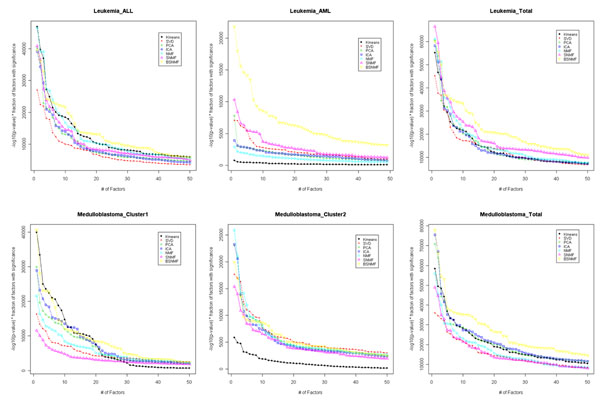
Results: Here we evaluated the clustering performance of orthogonal and non-orthogonal MFs by a total of nine measurements for performance in four gene expression datasets and one well-known dataset for clustering. Specifically, we employed a non-orthogonal MF algorithm, BSNMF (Bi-directional Sparse Non-negative Matrix Factorization), that applies bi-directional sparseness constraints superimposed on non-negative constraints, comprising a few dominantly co-expressed genes and samples together. Non-orthogonal MFs tended to show better clustering-quality and prediction-accuracy indices than orthogonal MFs as well as a traditional method, K-means. Moreover, BSNMF showed improved performance in these measurements. Non-orthogonal MFs including BSNMF showed also good performance in the functional enrichment test using Gene Ontology terms and biological pathways.
Conclusions: In conclusion, the clustering performance of orthogonal and non-orthogonal MFs was appropriately evaluated for clustering microarray data by comprehensive measurements. This study showed that non-orthogonal MFs have better performance than orthogonal MFs and K-means for clustering microarray data.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
